







这期分享一篇2024年5月发表于 Nat Commun (IF 14+)的文章,作者基于系统解剖肿瘤-正常单细胞生态系统横跨30种癌症类型的1000个肿瘤。
该文章使用桓峰基因公众号里面生信分享教程即可实现,有需要类似思路的老师可以联系我们!
摘 要
肿瘤微环境的复杂性对肿瘤治疗提出了重大挑战。在这里,为了全面研究肿瘤-正常生态系统,我们对来自 1070 个肿瘤和 493 个正常样本的 490 万个单细胞转录组进行了综合分析,并结合泛癌 137 个空间转录组,8887 个 TCGA 和 1261 个检查点抑制剂治疗的肿瘤。我们定义了构成肿瘤-正常生态系统的无数细胞状态,并确定了不同细胞类型和器官的标志性基因特征。我们的图谱描述了由 AKR1C1 或 WNT5A 标记的炎症成纤维细胞在细胞相互作用和空间共定位模式方面的区别。共现分析揭示了富含干扰素的群落状态,包括三级淋巴结构 (TLS) 成分,它们在肿瘤、邻近正常组织和健康正常组织之间表现出不同的重新连接。通过免疫疗法治疗的癌症 (n = 1261),包括我们的肺癌队列 (n = 497),证实了干扰素富集的社区状态对免疫疗法的有利反应。空间转录组的反褶积区分了免疫治疗有利成分中富集 TLS 和非富集的细胞类型。我们对肿瘤-正常生态系统的系统解剖提供了对肿瘤间和肿瘤内异质性的更深层次的理解。

文章研究思路及流程:
单细胞分辨率下泛癌肿瘤-正常景观的概述
(A)本研究中收集的30种癌症类型的scRNA-seq队列概述。
(B)肿瘤-正常单细胞转录组图谱和NMF处理流程。
肿瘤正常单细胞图谱子集的UMAP可视化,由(C)细胞类型和(D)器官来源着色。
E带有自动汤效应检测算法的NMF模块的图形聚类原理图及后续分析。
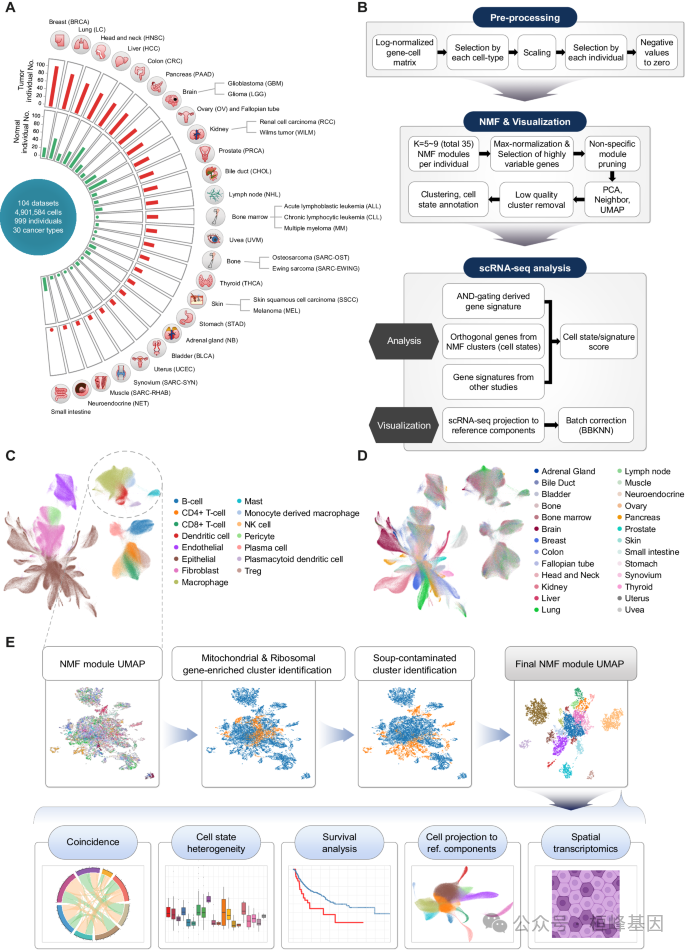
生信分析流程
数据集选择
单细胞数据:scRNA-seq datasets comprising 1070 tumors, 493 normal (adjacent normal + healthy normal) samples, and approximately 4.9 million cells from 999 individuals
转录组数据:28 cancer types TCGA RNA-seq,
immunotherapy-treated cohorts (4 cancer types, 8 cohorts)生信分析方法
根据文章的分析流程提取所有的分析内容,整理出来就17个分析条目,每个条目都包括分析的内容,这些分析构成了整个文章,本文基于单细胞转录组、bulk转录组分析结合多种实验结果的干湿类文章,下面我们就看看哪些分析可以利用桓峰基因公众号的教程来实现,点击分析条码就会跳转到对应公众号的教程,跟着教程做,您也能发轻松发高分,如下:
单细胞相关分析:
5.AND-gating 算法提取肿瘤富集或免疫治疗有利的基因特征
6.注释肿瘤特异性标记基因特征的生物学功能(Enrichr)
12.构建连接的网络(Gephi)
14.泛癌症空间转录组分析(cell2location)
17.绘图相关方法:
实验验证方法:
RNA smFISH
研究结果
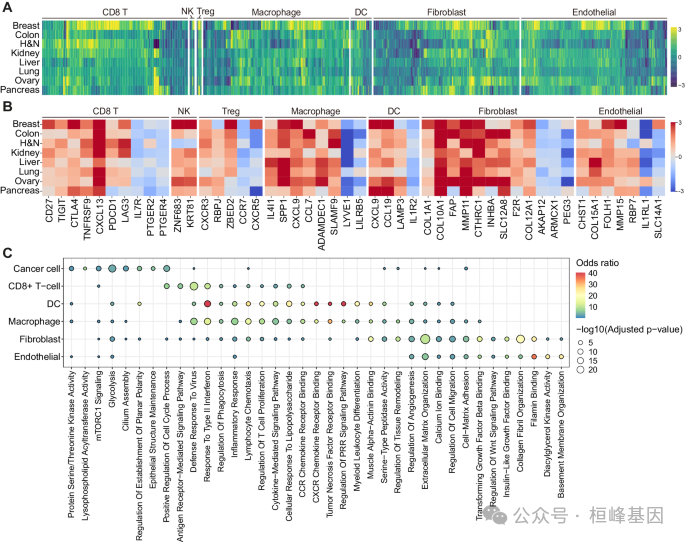
1. 跨器官组织生态系统标志基因景观
(A)跨器官肿瘤和正常生态系统中标志基因的表征
(B)在图2A中发现的跨越癌症和细胞类型的标志基因的详细热图。
(C)不同细胞类型肿瘤标志基因的基因本体分析。
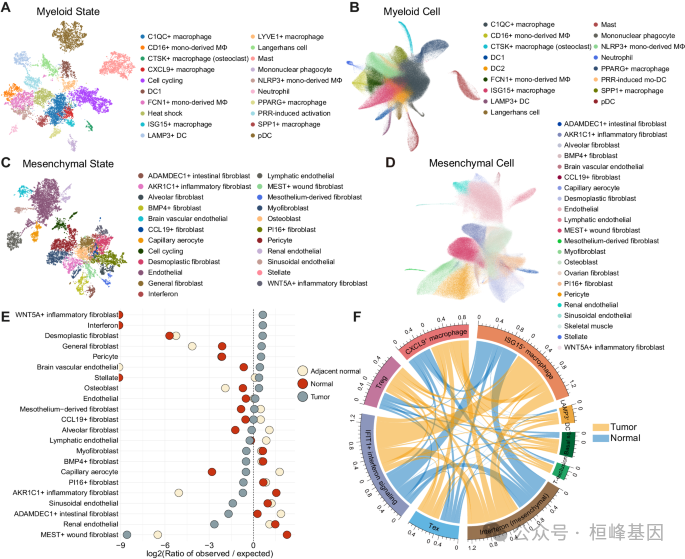
2. 将肿瘤-正常生态系统反卷积成异质细胞状态
(A)髓细胞状态的UMAP可视化。
(B)髓细胞相应参考成分分析。
(C)间充质细胞状态的UMAP可视化。
(D)间充质细胞相应参比成分分析。
(E)间充质细胞状态组织富集的Ro/E分析。
(F)Circos图显示正常和肿瘤组织中细胞状态的互作。
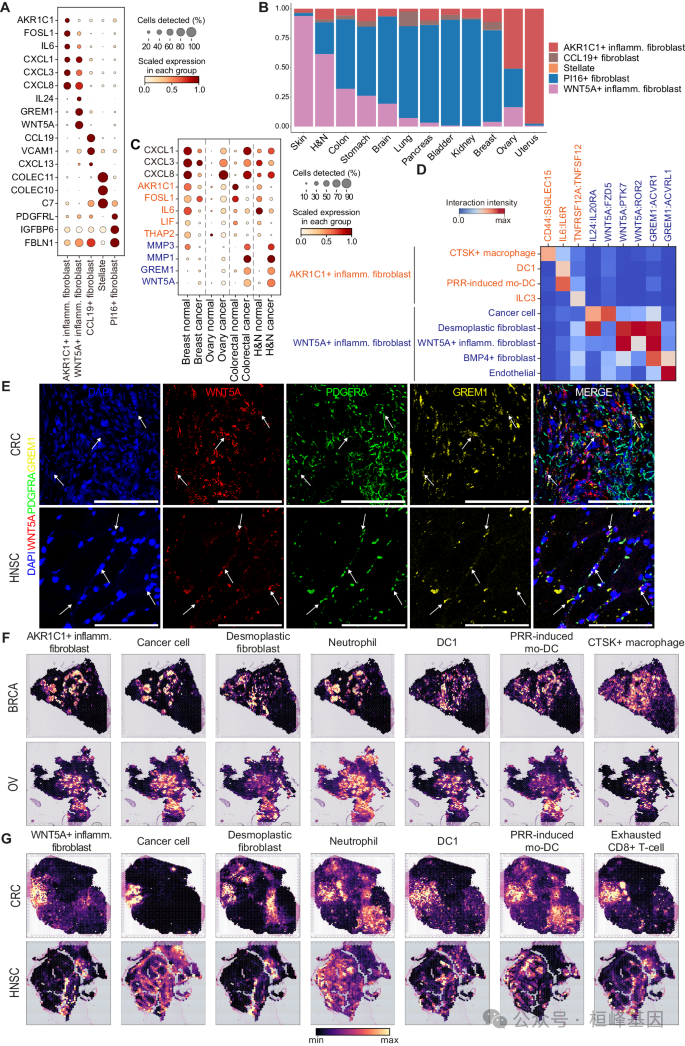
3. AKR1C1+和WNT5A+炎性成纤维细胞的表征
(A)炎性成纤维细胞亚型标记基因表达的点阵图。
(B)炎性成纤维细胞跨器官分布,其中y轴表示肿瘤组织中炎性成纤维细胞的比例。
(C)点图显示相关器官正常组织和肿瘤组织中AKR1C1+和WNT5A+炎性成纤维细胞标记基因的表达。
(D)AKR1C1+和WNT5A+炎性成纤维细胞与其他细胞类型的配体受体相互作用。
(E)CRC和HNSC组织间质中WNT5A、PDGFRA和GREM1原位RNA smFISH检测的代表性图像。
(F)AKR1C1+和(G)WNT5A+炎性成纤维细胞与相关器官其他细胞类型的空间共定位模式。
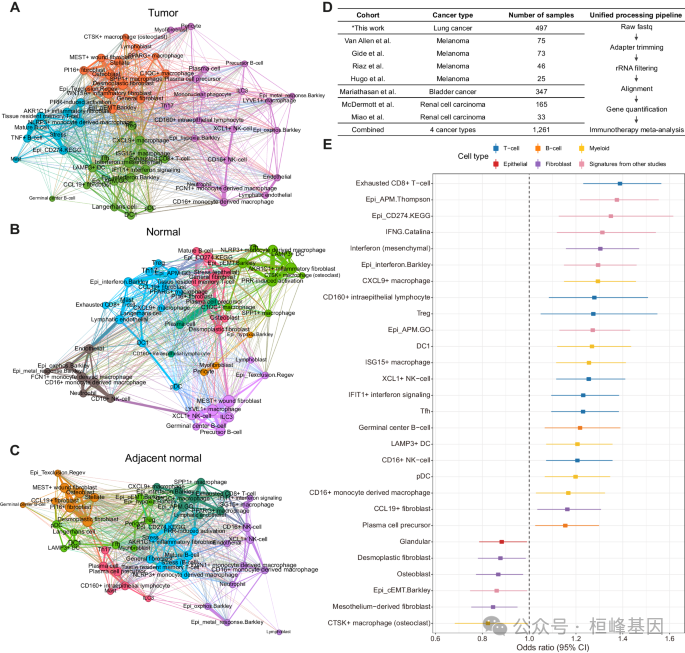
4. 肿瘤特异性干扰素富集和致瘤前社区的发生和免疫治疗预测细胞状态的测定
(A)肿瘤互作网络图。
(B)正常互作网络图
(C)邻近正常组织互作网络图。
(D)本研究中使用的免疫疗法治疗的大量RNA-seq队列的反应数据摘要。
(E)免疫治疗反应预测细胞状态和基因特征的森林图。
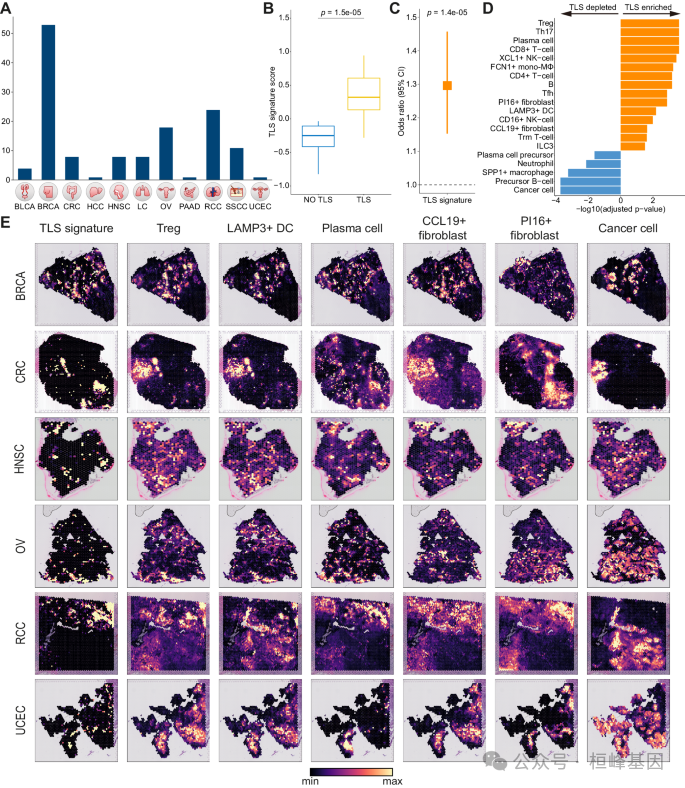
5. 跨多种癌症类型肿瘤生态系统的空间转录组分析
(A)本研究分析了泛癌症空间转录组的概况。
(B)TLS点与非TLS点的TLS签名得分对比箱线图。
(C)在接受免疫治疗的大量RNA-seq队列中,TLS签名对免疫治疗反应的预测能力。
(D)利用TLS标记的RCC空间转录组绘制富集tls的细胞类型Barplot。
(E)不同肿瘤细胞类型TLS信号的空间共定位。
Reference
Kang, J., Lee, J.H., Cha, H. et al. Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types. Nat Commun 15, 4067 (2024).
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出各种系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









