








论文:https://arxiv.org/abs/2205.07249
代码:https://github.com/pengxingang/Pocket2Mol
Pocket2Mol : 基于3D蛋白质口袋的高效分子采样
本文介绍由xingang peng等人发表于ICML上的文章:Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets。作者提出了一种新的可以满足口袋施加的多个几何约束的采样方法:Pocket2Mol,这是一个由两个模块组成的 E(3)-等变生成网络,它不仅可以捕获结合口袋原子之间的空间和键合关系,还可以在不依赖 马尔科夫链蒙特卡洛方法(MCMC)的情况下从易于处理的分布中以口袋表示为条件对新候选药物进行采样。其中,基于口袋药物设计改进如下: 1)开发一种新的深度几何神经网络来精确建模口袋的三维结构; 2)设计新的采样策略,实现更高效的条件三维坐标采样; 3)模型中对一对原子间化学键进行取样的能力的标志。实验结果表明,从 Pocket2Mol 中取样的分子具有明显更好的结合亲和力和其他药物特性,例如药物相似性和合成可及性。
介绍
早期的方法通过整合评估函数(如采样分子和口袋之间的对接分数)来修改无口袋模型,以指导候选搜索 1 ^1 1。另一种类型的模型将3D口袋结构转化为分子的SMILES字符串或2D分子图 2 ^2 2,而没有模拟小分子结构和3D口袋之间的相互作用。开发条件生成模型来模拟三维口袋结构中的三维原子密度分布,然后,这个问题的挑战点转移到了从学习分布中的结构采样算法的效率。此外,以往的模型过于强调原子的三维位置的重要性,而忽略了化学键的生成,导致实际中原子之间的连接不现实。
相关工作
基于三维蛋白质口袋的分子生成
- 一种改进的GAN模型来表示蛋白质口袋中隐藏空间中的分子,并使用字幕网络将这些表示解码为SMILES字符串。或者设计了两个结构描述符对口袋进行编码,并使用条件RNN生成了SMILES。
- 另一个工作思路开始考虑口袋和小分子的3D分子结构。提出了一种配体神经网络来生成3D分子结构,并利用蒙特卡洛树搜索来优化与特定口袋结合的候选分子。
基于向量特征的等变网络
通常采用基于gnn的架构来实现三维物体的全局旋转等方差。然而,它们要求每一层的输入和隐藏特征是等变的,这与每个氨基酸的侧链角等向量特征不相符。
原子位置的生成
- 一种常见的策略是预测新原子和所有先前原子之间的距离分布,并从节点分布中取样。
- 另一种策略是建立局部球坐标系,预测局部空间中的位置,但是欧氏空间和球面空间之间的转换效率低且不直接。
方法
Pocket2Mol的核心思想是根据已经存在的原子来了解口袋中每个位置的原子或键类型的概率分布。。为了了解这种特定于上下文的分布,作者采用自回归策略来预测训练药物的随机掩模部分。
生成过程
- 形式上,蛋白质口袋表示为一组带有坐标

2.生成的具有 n 个原子的分子片段表示为坐标

- 第 i 个重原子、它的配位和与其他原子的价键。模型记为φ,生成过程定义如下:

产生过程由四个主要步骤组成,如图一所示。
(1) 该模型的前沿预测因子 F f r o F_{fro} Ffro将预测当前分子片段的前沿原子。前沿被定义为可以共价连接到新原子的分子原子。如果所有的原子都不是前沿,则表明当前分子是完整的,生成过程终止。
(2) 其次,模型从边界集合中采样一个原子作为焦点原子。
(3) 然后,基于焦点原子,模型的位置预测器 f p o s f_{pos} fpos预测新原子的相对位置。最后,该模型的原子元素预测器 f e l e f_{ele} fele和键类型预测器 f b o n d f_{bond} fbond将预测具有现有原子的元素类型和键类型的概率,然后对新原子的元素类型和价键进行抽样。
(4) 这样,新原子就成功地加入到当前的分子片段中,生成过程一直持续到没有发现前沿原子为止。

模型结构
基于上述生成过程,模型需要由四个模块组成:编码器、边界预测器、位置预测器和元素和键预测器。、
E(3) - 等级神经网络
具有标量和向量特征的3D图中代表顶点和边缘可以帮助增强神经网络的表达能力。蛋白质口袋P(pro)和分子片段G(mol)n中的所有顶点和边都与标量和矢量特征相关联,以更好地捕捉三维几何信息。
对原始的GVP进行了修改,在GVP的输出向量上增加了一个矢量非线性激活,记作Gper:

编码器
将蛋白质口袋和分子片段表示为k近邻(KNN)图,其中顶点是原子,每个原子与其k近邻相连接。输入的矢量顶点特征包括原子的坐标,矢量边缘特征是三维空间中边缘的单位方向向量。
首先,利用多个嵌入层来嵌入顶点
(
v
i
(
0
)
,
v
e
c
v
i
(
0
)
)
(v^{(0)}_i, vec{v}^{(0)}_i)
(vi(0),vecvi(0))和边缘
(
e
i
j
(
0
)
,
e
⃗
i
j
(
0
)
)
(e^{(0)}_{ij},\vec{e}^{(0)}_{ij})
(eij(0),eij(0))特征。然后将L个消息传递模块
M
l
(
L
=
1
,
…
,
L
)
M_l(L = 1,…,L)
Ml(L=1,…,L)和更新模块
U
l
(
L
=
1
,
…
,
L
)
U_l(L = 1,…,L)
Ul(L=1,…,L)交错连接,学习局部结构表示

消息传递模块形式为:

矢量消息的计算方法是将顶点和边的矢量特征与标量特征相乘后求和,实现顶点和边之间、标量特征和矢量特征之间的信息交互。更新模块形式为:

预测
边界预测: 将几何向量MLP (GV-MLP)定义为一个GVP块后跟一个GVL块,称为
G
m
l
p
G_{mlp}
Gmlp。前沿预测器以原子i的特征为输入,利用一个GV-MLP层来预测前沿
p
f
r
o
p_fro
pfro的概率,如下所示:

位置预测指标:
位置预测器以焦点原子
i
i
i的特征为输入,预测新原子的相对位置。由于矢量特征在模型中是等变的,它们可以直接生成指向焦点原子坐标
r
i
r_i
ri的相对坐标
Δ
r
i
\Delta r_i
Δri。将位置预测器的输出建立为具有对角协方差的高斯混合模型
p
(
Δ
r
i
)
=
∑
k
=
1
K
π
i
(
i
)
N
(
u
i
(
k
)
,
Σ
i
(
k
)
)
p(\Delta r_i) =\sum^{K}_{k=1}\pi^{(i)}_iN(u^{(k)}_i,\Sigma^{(k)}_i)
p(Δri)=∑k=1Kπi(i)N(ui(k),Σi(k))其中,多个神经网络对参数的预测如下:

元素和键预测器: 在预测新原子
i
i
i的位置后,元素-键预测器将预测新原子
i
i
i的元素类型以及现有分子片段中所有原子之间的价键
q
(
∀
q
∈
V
(
m
o
l
)
)
q(\forall q \in V^{(mol)})
q(∀q∈V(mol))。图2展示了预测神经网络的结构。

首先,我们在所有原子中收集
k
k
k近邻原子
j
∈
K
N
N
(
i
)
j \in KNN(i)
j∈KNN(i),然后利用一个消息传递模块,将来自邻居原子的局部信息集成到新的原子
i
i
i位置,作为其表示
(
v
i
,
v
⃗
i
)
(v^i,\vec{v}^i)
(vi,vi),据此预测原子
i
i
i的元素类型。
在并行路径中,原子
i
i
i和
q
q
q之间的边表示为
(
z
i
q
,
z
⃗
i
q
)
(z_{iq},\vec{z}_{iq})
(ziq,ziq),是原子
i
i
i的特征、原子
q
q
q的特征和边
e
i
q
e_{iq}
eiq的处理特征的拼接,然后是一个GV-MLP块,即:

其中
(
e
i
q
′
,
e
⃗
i
q
)
(e'_{iq}, \vec{e}_{iq})
(eiq′,eiq)为边缘嵌入处理后的输入边缘特征和一个GV-MLP块。
对于向量特征,提出了一种新的注意模块,定义如下

训练
在训练阶段,随机屏蔽分子中的原子,训练模型恢复被屏蔽的原子。具体来说,对于每一对口袋配体,从均匀分布的U[0,1]和掩模对应的分子原子数中取样一个掩模比。剩下的与被掩盖原子有价键的分子原子被定义为边界。然后,位置预测器和元素-键预测器试图通过预测与边界相对应的位置、元素类型和与剩余分子原子的键来恢复与边界具有价键的被掩盖原子。如果所有的分子原子都被掩蔽,边界被定义为4a内具有掩蔽原子的蛋白质原子,边界周围的被掩蔽原子将被恢复。对于元素类型预测,我们在查询位置添加了一个表示Nothing的元素类型。在训练过程中,我们不仅对用于元素类型预测的掩模原子的位置进行采样,还对来自环境空间的负位置进行采样,并将它们的标签赋值为Nothing。
前沿预测的损失
L
f
r
o
L_{fro}
Lfro是预测前沿的二元交叉熵损失。位置预测器的损失
L
p
o
s
L_{pos}
Lpos是被掩盖原子位置的负对数似然。对于元素类型和键类型的预测,我们使用交叉熵损失进行分类,分别表示为
L
e
l
e
L_{ele}
Lele和
L
b
o
n
d
L_{bond}
Lbond。
整体损失函数是上述四种损失函数的总和

采用Adam优化器同时对编码器和所有三个预测器进行优化。
结果

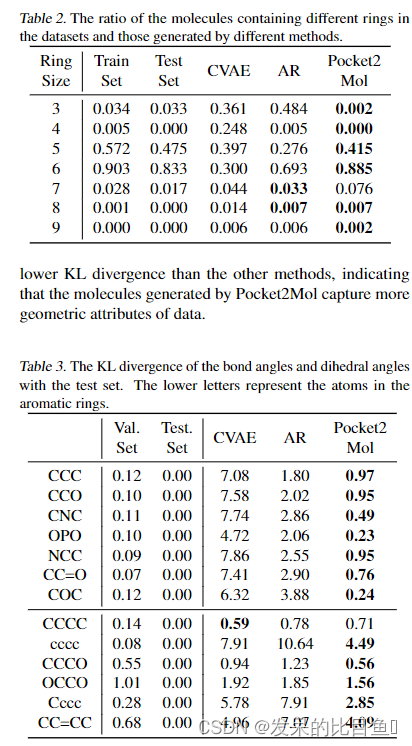

Pocket2Mol,这是一个由图神经网络组成的E(3)等变生成网络,用于建模三维蛋白质口袋的化学和几何特征,并采用一种新的高效算法来采样基于口袋条件的新的3D候选药物。实验表明,Pocket2Mol生成的分子不仅具有更好的亲和性和化学性质,而且具有更真实和准确的结构。
参考
- Structure-based de novo drug design using 3d deep generative models
- From target to drug: Generative modeling for the multimodal structure-based ligand design. Molecular Pharmaceutics
- novo molecule design through the molecular generative model conditioned by 3d information of protein binding sites.
















 662
662











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











