






欢迎关注"生信修炼手册"!
通过基因相关注释,可以知道变异位点在基因组上的位置和对蛋白质编码的影响。在进行注释之前,首先需要下载物种对应的数据库,以human为例,命令如下
annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
下载成功后,humandb的文件列表如下
├── annovar_downdb.log
├── hg19_refGeneMrna.fa
├── hg19_refGene.txt
└── hg19_refGeneVersion.txt数据库准备好之后,就可以进行注释了,命令如下
annotate_variation.pl —geneanno -buildver hg19 ex1.avinput humandb
运行过程中的log信息如下
NOTICE: Output files were written to ex1.avinput.variant_function, ex1.avinput.exonic_variant_function
NOTICE: Reading gene annotation from humandb/hg19_refGene.txt ... Done with 63481 transcripts (including 15216 without coding sequence annotation) for 27720 unique genes
NOTICE: Processing next batch with 21 unique variants in 21 input lines
NOTICE: Reading FASTA sequences from humandb/hg19_refGeneMrna.fa ... Done with 15 sequences
WARNING: A total of 405 sequences will be ignored due to lack of correct ORF annotation会输出两个文件,后缀分别为.variant_function和.exonic_variant_function。
1. variant_function
这个文件在输入文件的前面,新加了两列,第一列代表变异位点在基因上的区域,比如外显子,内含子,基因间区等;第二列给出对应的基因。示例如下
UTR5 ISG15(NM_005101:c.-33T>C)
UTR3 ATAD3C(NM_001039211:c.*91G>T)
intronic DDR2
intronic DNASE2B
intergenic UBIAD1(dist=43968),DISP3(dist=135699)
exonic IL23R
exonic ATG16L1annovar将基因组划分成了9种区间
exonic
splicing
ncRNA
UTR5
UTR3
intronic
upstream
downstream
intergenic
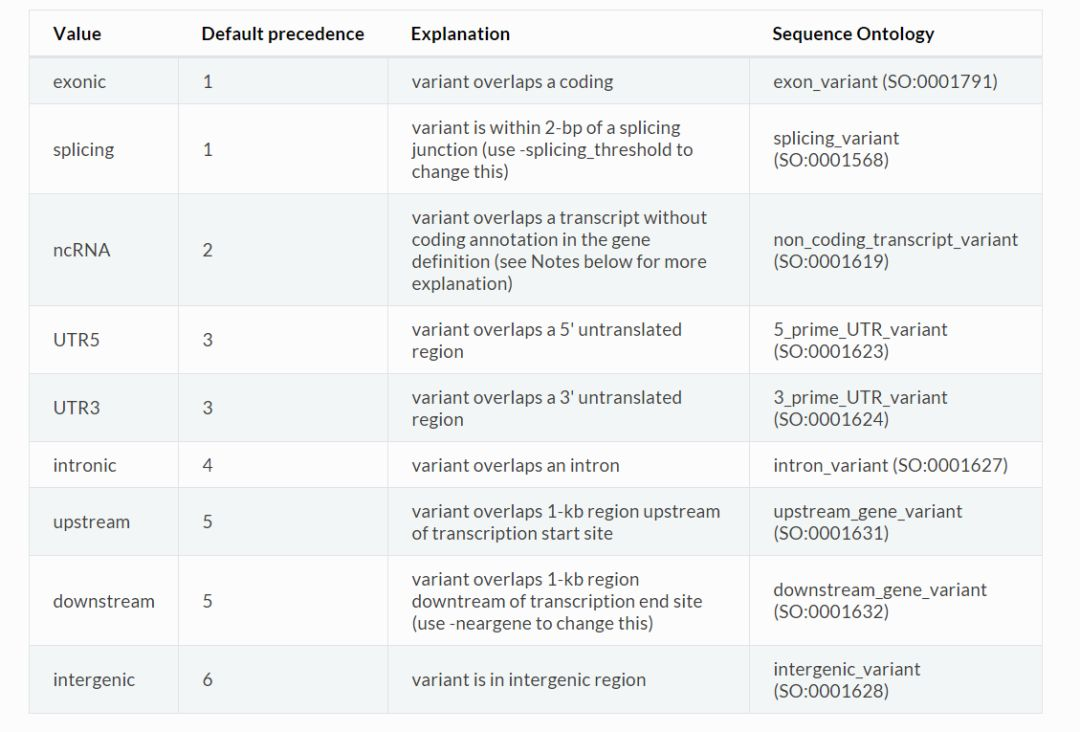
exonic特指编码蛋白的外显子区;UTR5和UTR3特指不翻译蛋白的外显子区;splicing指的是位于内含子边界(默认2bp以内)的区域;ncRNA指的是非编码蛋白的基因区域;intronic指的是内含子区;upstream指的是转录起始位点上游1Kb以内的区域;downstream指的是转录终止位点下游1kb以内的区域;intergenic值的是基因间区。
在判断一个变异位点所处区域时,以上9种区间的优先级是不同的,下图中列出了每种区间的优先级,数字越小,优先级越高。
如果一个变异位点位于某个基因区域时,第二列会给出对应的基因名称,如果有多个基因名称,则逗号分隔,比如
exonic ATG16L1
如果一个变异位点位点不在基因区域,第二列会给出上下游最近的基因的名字和距离,比如
intergenic UBIAD1(dist=43968),DISP3(dist=135699)
2. exonic_variant_function
这个文件只对位于exonic区间的变异位点,给出对应的氨基酸变化信息。在输入文件的基础上新增了3列,第一列代表行数,第二列代表变异类型,第三列代表氨基酸的变化情况,示例如下
line9 nonsynonymous SNV IL23R:NM_144701:exon9:c.G1142A:p.R381Q,annovar提供了以下几种变异类型
frameshift insertion
frameshift deletion
frameshift block substitution
stopgain
stoploss
nonframeshift insertion
nonframeshift deletion
nonframeshift block substitution
nonsynonymous SNV
synonymous SNV
unknown
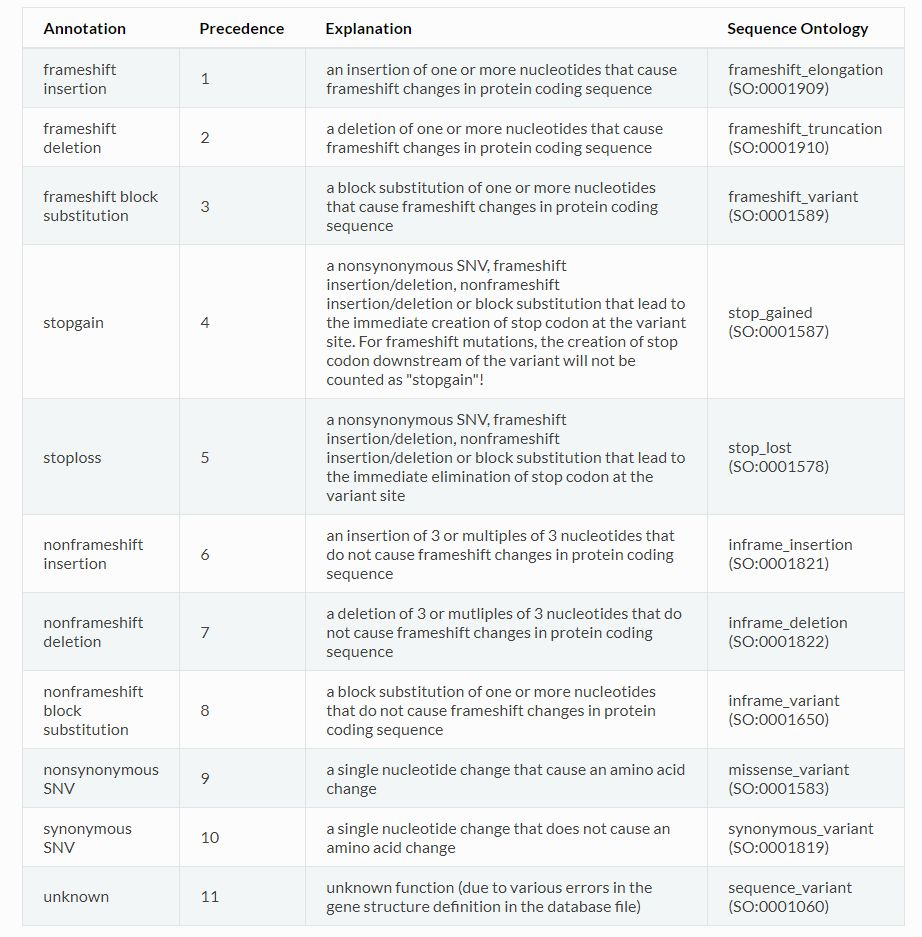
在定义变异类型时,首先基于4种基本的变异类型,SNV, insertion, deletion, block substitution, 再结合其对蛋白编码的影响。对于SNV而言,引起了蛋白质变化的就是synonymous SNV, 蛋白质没有变化的就是
nonsynonymous SNV;对于剩下的3种基本变异类型,在考虑对蛋白质的影响时,分为了移码frameshift和非移码nonframeshift 两种。stopgain指的是突变之后,原本的密码子变成了终止密码子,stoploss指的是突变之后,原本的终止密码子变成了普通密码子,导致翻译情况变化较大。unknown代表不清楚该变异对蛋白的影响。
和分析变异位点所处区间类似,评估变异类型时也有优先级的区分,优先级如下
在表示蛋白质的影响时,annovar采用的是自己定义的表示规则,如果想要使用HGVS定义的规则,只需要在运行时添加-hgvs参数,示例如下
annotate_variation.pl —geneanno -buildver hg19 -hgvs ex1.avinput humandb
添加这个参数之后,exonic_variant_function文件的第三列示例如下
IL23R:NM_144701:exon9:c.1142G>A:p.R381Q
可以看到,采用的是HGVS的命名方式。
在使用annovar注释时,还有一个小技巧。因为只需要输入文件的前5列,当我们只有基因区间文件,比如bed格式的文件时,可以将4,5列用0填充,这样的格式annovar也是可以识别的,这样就可以对基因组上的区间进行基因相关的注释了。
扫描关注微信号,更多精彩内容等着你!


















 1030
1030

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









