






Yuan, H., Kelley, D.R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat Methods 19, 1088–1096 (2022). https://doi.org/10.1038/s41592-022-01562-8
https://github.com/calico/scBasset![]() https://github.com/calico/scBasset
https://github.com/calico/scBasset
摘要
1. 由于高维和稀疏的特性,scATAC数据的分析一直面临很多挑战。
2. scBasset是一个 建模scATAC数据 的 sequence-based 卷积神经网络方法
3. scBasset利用潜在可及性peaks 的DNA序列信息 在scATAC和单细胞多组学数据的多种任务上均实现了最好的表现。如 细胞聚类、scATAC谱降噪、数据整合(with scRNA)、TF活性推断。
介绍
1. 有很多解决scATAC数据高维和稀疏性的方法,大致分为:sequence-free 和 sequence-dependent。
前者通常是生成稀疏的peak-by-cell矩阵,忽视DNA序列,如对矩阵进行线性转换的PCA和latent semantic indexing,将细胞投射到一个低维空间(压缩peaks)。SCALE和cisTopic使用Latent Dirichlet Allocation 或 变分自编码器。这些方法能够检测生物学上有意义的协方差,从而有效 represent 以及cluster 或 classify 细胞。也就是更有效的标识细胞身份。------然而它们忽视了序列信息,并依赖于事后的 motif-matching工具 来将 可及性与TF联系起来。
后者常见chromVAR,BROCKMAN,通过TF motif 或 k-mer含量来表示peaks ,并聚集peaks中的这些特征,来学习细胞的表示。
而作者基于 DNA 序列的深度卷积神经网络 (CNN),为 scATAC 提出了一种更具表现力的sequence-dependent模型,其中,初始卷积层学习了TF motifs和其它序列factors。后续层计算这些特征的非线性组合,以产生序列的明确embeddings,然后最后的线性层将序列embeddings转换为可及性的预测。这些线性层参数的矩阵能够对多种细胞进行明确的embed,依赖于它们如何利用序列embeddings的潜在变量。
scBasset是对Basset deep CNN架构的扩展,目的是从DNA序列预测单个细胞的染色质可及性。
结果
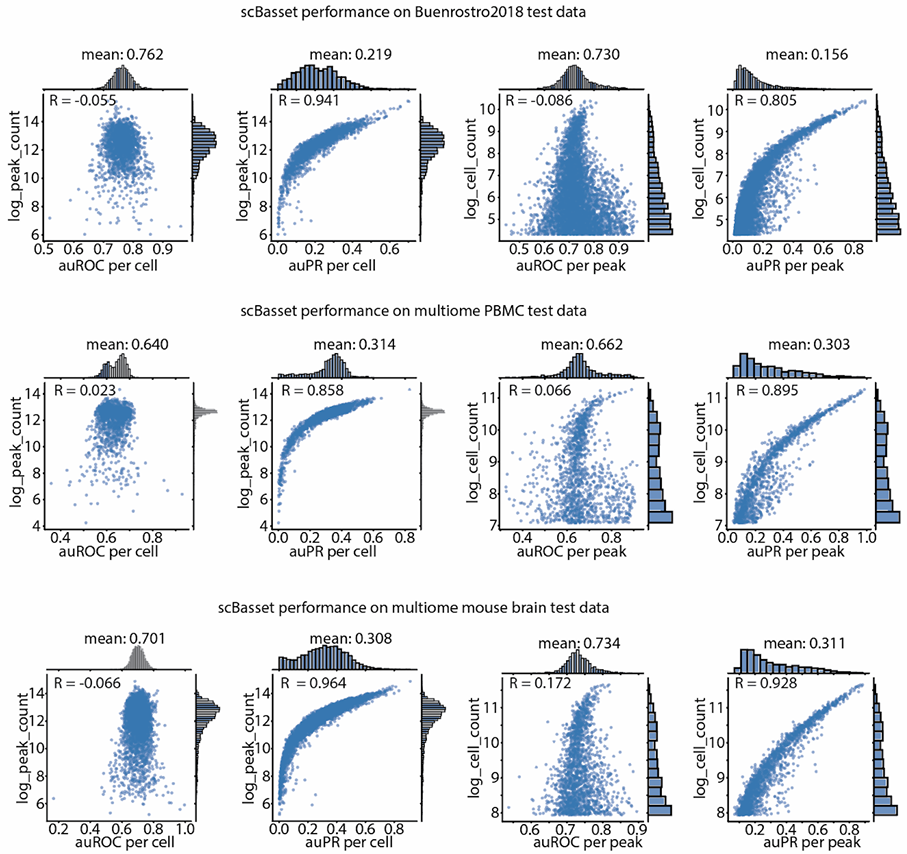
1. scBasset预测在held-out peaks(test_split by peaks)上的可及性
CNN是用来生成peaks_embeddings的,线性层转换是用来生成多个细胞的可及性的。意思是CNN层在所有单个细胞中的参数都是共享的??
(是的,因为它是一个多任务模型【shared-bottom,不同任务间共用底部的隐层】,模型的前部分(即从输入层到瓶颈层之前的所有层)对所有细胞的数据执行相同的操作,学习从序列数据中提取所有细胞普遍适用的特征。这种设计允许模型利用所有细胞的数据来学习表示,而不是为每种细胞独立学习一套参数。只有在瓶颈层之后的部分,即最终输出层,参数是特定于任务的。这是因为每个任务(region在每种细胞的可及性预测)可能需要基于提取的通用特征的不同贡献来做出决策。)
可以简单将最后一层线性层理解为:每个潜在变量代表多种调控元素,最后一层线性转换就是明确 每个细胞有多依赖这个调控元素。
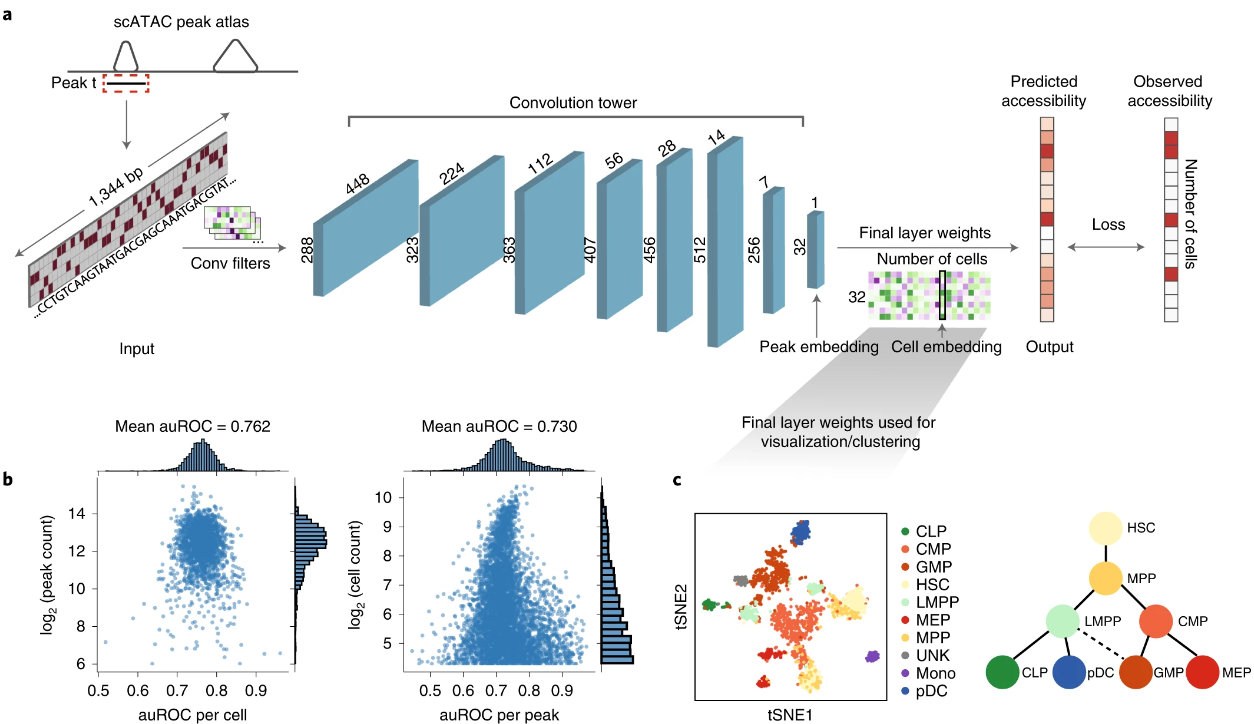
建议先用10x CellRanger scATAC pipeline预处理数据,得到raw peak-by-cell 二值count矩阵
输入长度:1344bp
8个 卷积block(1d 卷积-- batchnorm--maxpooling--GELU)
binary cross-entropy & stochastic gradient descent
数据集:
① Buenrostro2018 --2000个细胞---流式分选的造血分化细胞
② PBMCs --3000细胞--10x scRNA+scATAC外周血单核细胞
③ mouse brain PBMCs --5000细胞--10x scRNA+scATAC小鼠大脑细胞
①提供了细胞类型labels的ground-truth
评价:
① per cell实际上就是只考虑这一个细胞的所有peaks预测值,并与真实label比较后选出能够使其准确率最高的信号阈值
② 为了评估细胞类型的特异性,他才继续算了per peak,也就是这个peak在所有细胞中 是否能显示出差异
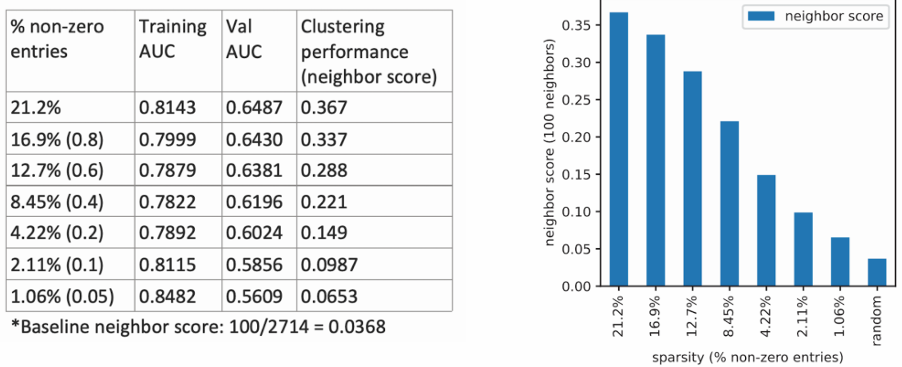
为了进一步评估测序深度的影响,对 10x multiome PBMCs 进行了不同程度的下采样,并训练了 scBasset。validation auROC 和细胞嵌入指标会随着测序深度的降低而降低,但即使数据集只包含 1% 的非零数据,scBasset 的性能仍优于随机数据集。
2. scBasset 最后一层 学习了细胞表示
2.1 将最后一层密集层 学习到的权重矩阵 作为单细胞的低维表示。
首先需要确保 细胞表示 与 测序深度 无关 ??怎么保证的?
2.2 聚类
Buen2018的细胞类型真实label,scBasset在多种测量下 表现更好。
label score:building a nearest-neighbor graph based on cell embeddings and asked what percentage of each cell’s neighbors share its same label
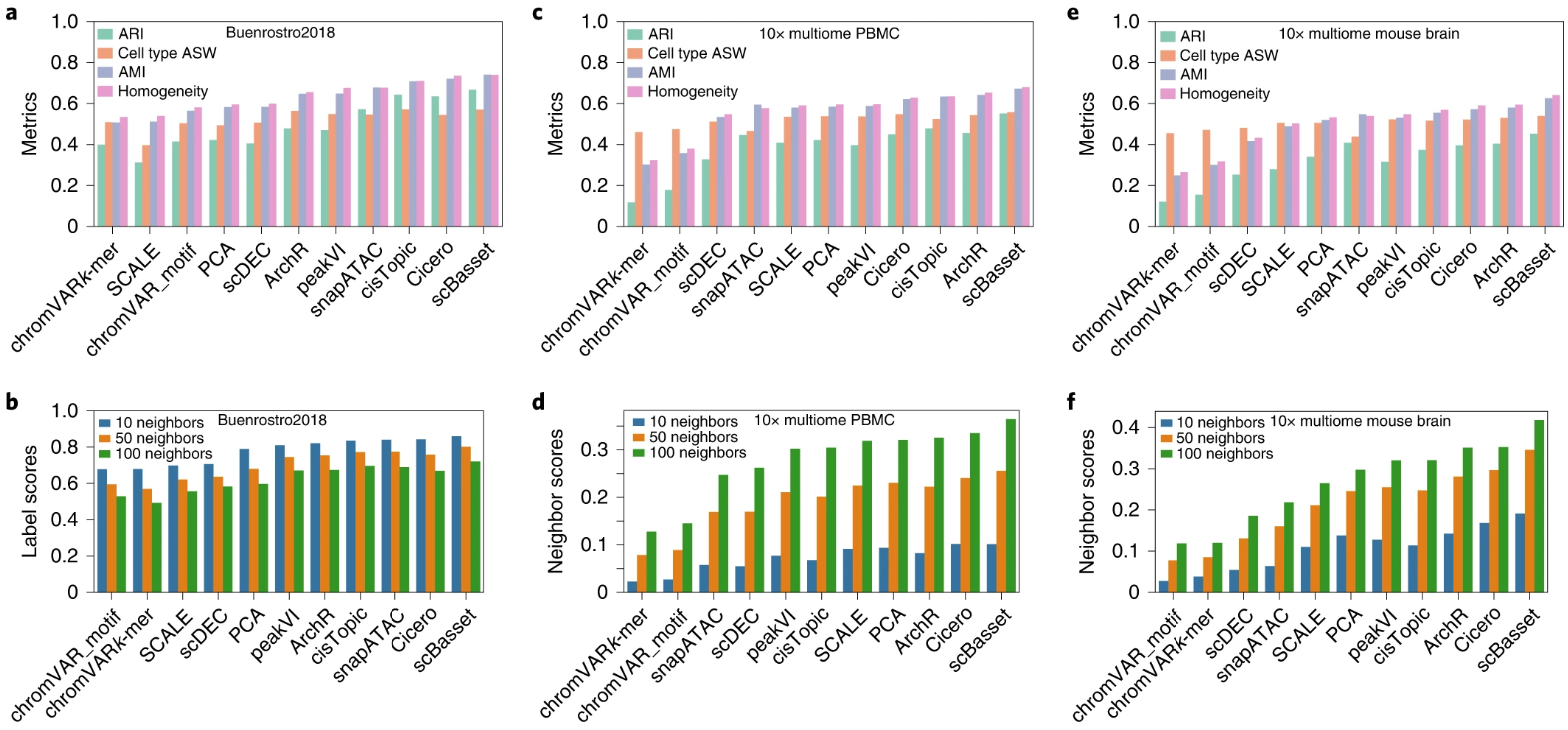
对于另外两组数据,没有细胞类型的真实label,因此As the ground-truth cell types for the multiome datasets are unknown, we used cluster identifiers from scRNA-seq Leiden clustering as cell type labels.
neighbor score:built independent nearest-neighbor graphs from the scRNA and scATAC and asked what percentage of each cell’s neighbors are shared between the two graphs
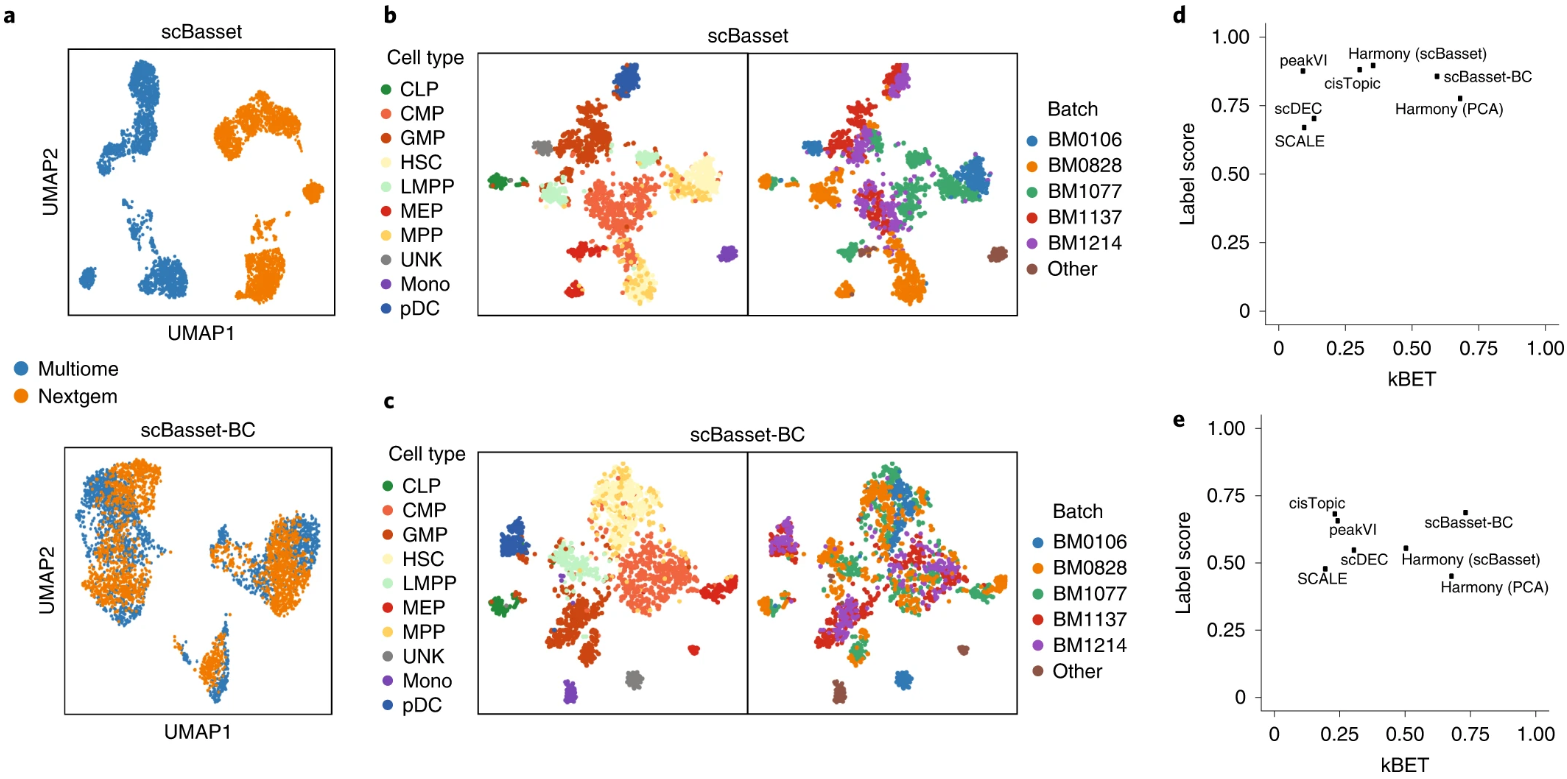
3. Batch-conditioned scBasset corrects cell embeddings for batch
批处理条件下的 scBasset 对 细胞嵌入进行 批次校正
3.1 不同donors导致的批次效用
在最后一个线性层即bottleneck之后,增加第二个全连接层,以预测特定批次对可及性的贡献。
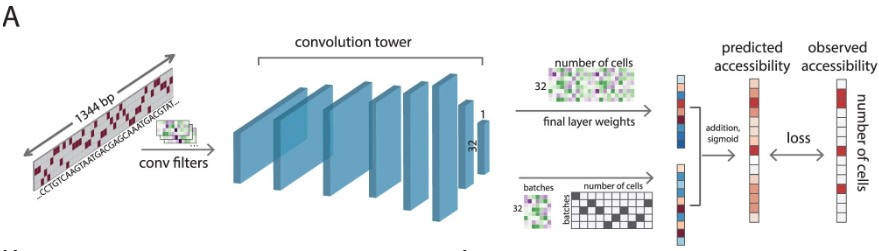
预计新添加的线性层将捕捉到 批次特异性变异,而原始权重矩阵将侧重于生物学相关变异的部分
通过在细胞特定层上引入 L2 正则项,可以控制信息流和批次混合程度。
3.2 scBasset-BC
对来自10x multiome 和 10x next GEM chemistries 的 PBMC scATAC进行混合,然后应用scBasset-BC来矫正其批次效应。用iLISI 和 kBET来评价混合表现,并用label score来量化生物学变异的保守性。与其它去批次效应的方法相比,scBasset-BC和 Harmony实现了batch mixing 和 生物学变异保守性 方面最好的平衡。
ad-PBMC;bce-Buen2018;
4. scBasset denoises single cell accessibility profiles降噪
稀疏性--很多假阴性,通常很多方法 会根据cells进行累积。
在Buen2018数据中 采样500peaks 和 200 cells,并直接进行可视化----raw cell-by-peak matrix,以及经过scBasset预测后的即降噪的matrix。raw中cells和peaks根据测序深度聚类,显示没有生物学相关的模式。而scBasset降噪后,相同细胞类型的细胞共享相似的可及性谱,细胞的分层聚类与ground-truth label十分吻合。
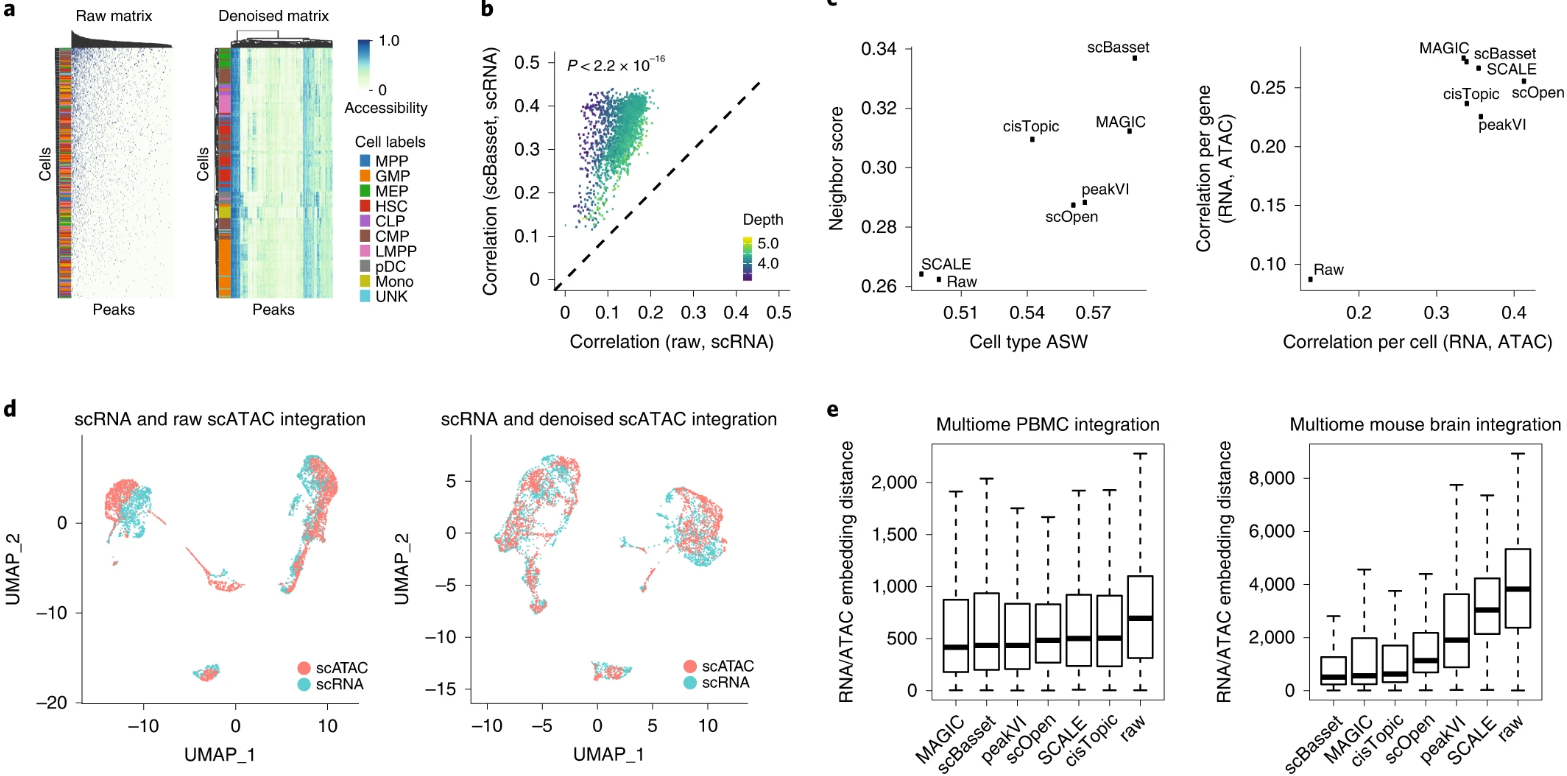
4.1 评估scBasset在Buen2018上的去噪性能,以及去噪后对 cell-cell距离估计 和 cell embed 的影响。
有效的去噪将提高 基因可及性估算值 与 基因RNA 表达量之间的相关性。测序深度更小的peaks改进越大。gene accessibility and expression。scRNA是用scVI降噪的,scATAC是用scBasset。
computed accessibility scores for each gene by averaging the predicted accessibility values at all promoter peaks before and after denoising
4.2 将独立的scRNA和scATAC谱整合到一个共享的 latent space,是许多scATAC注释和分析的关键步骤。scBasset去噪的scATAC会提升其整合的表现。
将10x multiome的scRNA和scATAC视为独立的数据,定量测量对于每一个匹配的细胞的RNA和ATAC embeddings 之间的rank distance。
scBasset and MAGIC outperformed alternative methods for data integration
5. 在单细胞分辨率下推断TF活性
向训练好的 scBasset 模型提供了含有和不含有特定 TF motif的合成 DNA 序列(二核苷酸shuffled peak),并根据预测的可及性变化评估了该motif在每个细胞中的活性。如果某种 TF 在特定细胞中发挥激活作用,我们就会发现插入 TF motif 后,细胞的可及性增加。
插入了733 human CIS-BP motifs
5.1 系统比较scBasset与其它定量TFmotif活性的工具,如chromVAR,用10x PBMC multiome数据,RNA-seq 中测得的 TF 表达量可作为其motif活性的近似物
对于positive TF expression–activity correlation,scBasset预测的关联性显著高于chromVAR
对于negative TF expression–activity correlation,scBasset预测的关联性显著低于chromVAR
5.2 对于PBMC细胞类型的关键调控子来说,scBasset 相比chromVAR来说,TF 活性有更高的细胞类型特异性,并与TF表达更相关。
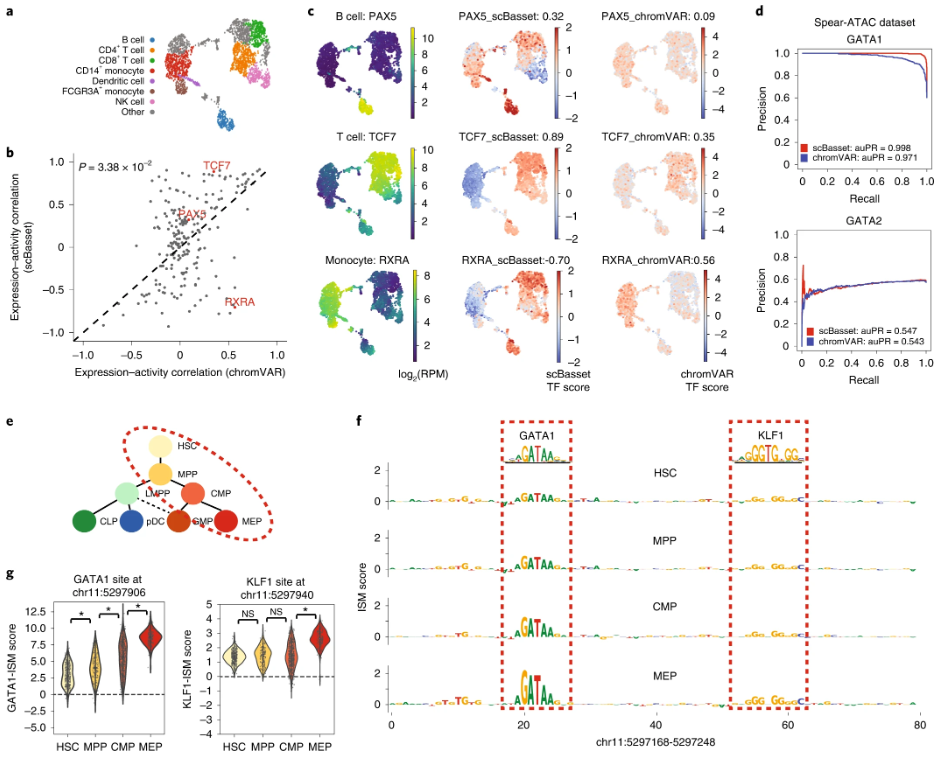
5.3 spear-ATAC:they targeted specific TFs with CRISPRi in single cells and read out the perturbed ATAC-seq profile。通过研究 TF 受到扰动的数据来验证 TF 的活动推断.
比较了 scBasset 和 chromVAR 的单细胞 TF 活性推断得分,即它们分别利用推断出的 GATA1 和 GATA2 分数,区分 sgGATA1 细胞和 sgNT 细胞以及 sgGATA2 细胞和 sgNT 细胞的能力。
所以这部分用了两种方式来说明scBasset对TF 活性推断的准确性,一种是利用插入motifs后可及性增加与TF表达之间的正负相关性;一方面是通过TF的敲除与否,分别预测出的该TF活性,来区分单个细胞(状态)的能力。
5.4 infer TF activity at per-cell per-nucleotide resolution
研究了β-globin基因的一个已知增强子(调控红细胞特异性β-globin的表达)。对其100 bp 序列的每个位点都进行了硅饱和诱变 (ISM)。
ISM score:位点突变后scBasset预测的可及性变化值。并对红细胞系中的每种细胞类型的ISM 分数做了平均。计算已知的该细胞类型的PWM分数,与预测的ISM分数 之间的PCC ,影响最大的核苷酸对应于 GATA1 和 KLF1 motif,众所周知,它们能与该增强子区域结合,并调控β-globin的表达。
scBasset 以单细胞分辨率学习可及性调控语法,可用于识别调控单个细胞和细胞系中 特定增强子的TF。
讨论
1/ sequence-dependent方法的局限性:参考基因组未考虑样本的变异,如CNV。我们的模型假设调控模式及其相互作用在整个基因组中具有普遍性,在某些基因组位点上可能不正确,这些位点上,进化导致了特殊的调控模式(雌性X染色体失活)
2/ 对于细胞状态的连续轨迹等应用,以及分散聚类可能会丢失信息的情况,单细胞分辨率可能是理想的选择。
3/ 展望:
可以同时对序列和细胞 进行小批量采样;
TFMoDISco可以应用于scBasset的ISM分数计算,从头发现motifs;
NN可以考虑序列信息,改善scATAC的peak calling;
迁移学习,pretrain-finetune
数据
10x配套scATAC+scRNA,pbmc_3k:
Datasets -Single Cell Multiome ATAC + Gene Exp. -Official 10x Genomics Support![]() https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/2.0.0/pbmc_granulocyte_sorted_3k10x配套scATAC+scRNA,小鼠的e18_5k :
https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/2.0.0/pbmc_granulocyte_sorted_3k10x配套scATAC+scRNA,小鼠的e18_5k :
Datasets -Single Cell Multiome ATAC + Gene Exp. -Official 10x Genomics Support![]() https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/2.0.0/e18_mouse_brain_fresh_5k两个10x multiome的scRNA数据是用scVI v.0.6.5进行处理的,降噪!聚类并根据marker genes生成cell type label!
https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/2.0.0/e18_mouse_brain_fresh_5k两个10x multiome的scRNA数据是用scVI v.0.6.5进行处理的,降噪!聚类并根据marker genes生成cell type label!















 1439
1439











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









