






 本文介绍了多个用于药物靶标预测和研究的在线数据库和工具,如TDRTargets、DrugMiner、TTD、TargetHunter、PharmMapper、ChemMapper、SEA、RFQSAR、SwissTargetPrediction、Anglerfish、SuperPred、COVIDep、idTarget、GalaxySagittarius和MolTarPred。这些资源提供基因组学、化学信息和药物活性数据,利用机器学习和结构相似性方法预测药物与靶标的相互作用,支持药物发现和新药开发研究。
本文介绍了多个用于药物靶标预测和研究的在线数据库和工具,如TDRTargets、DrugMiner、TTD、TargetHunter、PharmMapper、ChemMapper、SEA、RFQSAR、SwissTargetPrediction、Anglerfish、SuperPred、COVIDep、idTarget、GalaxySagittarius和MolTarPred。这些资源提供基因组学、化学信息和药物活性数据,利用机器学习和结构相似性方法预测药物与靶标的相互作用,支持药物发现和新药开发研究。
01 TDR Targets
这个数据库通过收集疾病病原体的各种基因组学和化学数据信息,实现对药物和靶标相互作用的鉴定和排序,包含 11 种细菌和真核生物的病原体的数据信息,以及超过 800,000 种生物活性化合物的数据信息。它既可以作为一个网站查找有关靶标、药物和/或感兴趣的生物活性化合物的信息,也可以作为对全基因组中的靶标进行优先排序的工具。
02 Drug Miner
这是一个用于药物靶点预测和功能注释的在线数据库和引擎。所有靶标均由机器学习算法 DrugMiner 预测,该预测算法的开发基于数千种药物-蛋白质的相互作用。值得注意的是,DrugMiner 仅预测人类药物靶标。最新版本中,用户可以提供蛋白Uniprot ID、名称或序列来自定义寻靶工作。
03 TTD
浙江大学药学院朱峰教授等人开发的药物靶标数据库TTD是全球第一个提供免费药物靶标信息的在线数据库,是药物靶标发现和新药开发领域具有国际影响力的数据平台。该数据平台的年访问量超过15万人次,总访问量达240余万人次,年平均被引次数超过200次。目前,该数据库收录了3500余个药物靶点,近4万个药物分子。旨在提供文献中所述的已知治疗蛋白和核酸靶标、靶向性疾病、通路信息以及针对每个靶标的相应药物/配体的信息。每个条目可以通过多种方法检索,包括目标名称、疾病名称、药物/配体名称、药物/配体功能和药物治疗分类。

04 TargetHunter
这个网站通过探索最大的化学基因组数据库 ChEMBL,实现了一种新颖的计算机靶标预测算法,即与其最相似的对应物相关的靶标。ChEMBL子集上前3的预测准确度达到 91.1%,优于已发布的算法及多分类模型。TargetHunter 还具有嵌入式工具 BioassayGeoMap,允许用户搜索可通过实验验证预测的生物靶标或脱靶的潜在靶标。它显著提高了化学基因组学研究人员在计算机药物设计和发现方面的生产力。
05 PharmMapper
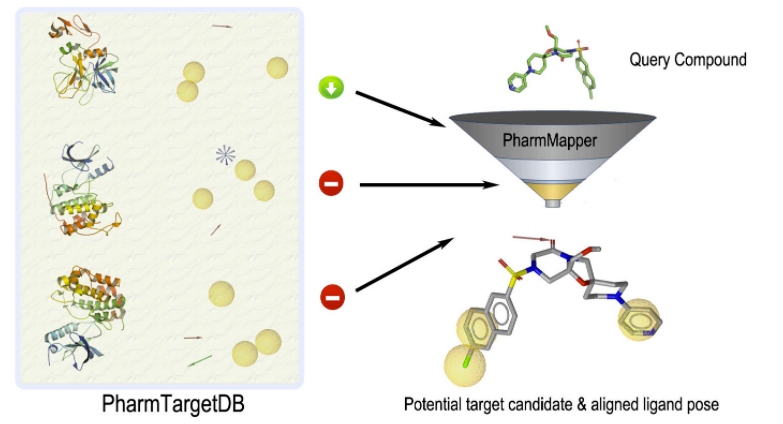
Pharmacmapper Server是华东理工大学李洪林教授团队开发的一个可以自由访问的在线服务器,旨在通过药效团映射的方法,为给定的探针小分子(药物、天然产物或其他新发现的结合靶标不明的化合物)识别潜在的靶标。受益于高效和稳健的映射方法,PharmMapper具有高通量的能力,可在数小时内从数据库中识别潜在的靶标候选对象。有一个庞大的内部药效团数据库(7000多个基于受体的药效团模型,涵盖1627个药物靶标信息)作为支持,PharmMapper可从TargetBank、DrugBank、BindingDB和PDTD中提取所有靶标。它将用户上传的分子(Mol2或SDF格式)与PharmTargetDB中的所有靶标进行最佳映射,并输出前N个潜在药物靶标以及相应分子的对齐构象 。
06 ChemMapper
ChemMapper跟pharmapper均出自李洪林教授团队,基于的理论是具有高度3D相似性的化合物可能有相对相似的靶标。它整合了不同来源的近30万个化学结构和药理学注释。总化合物超过300万。ChemMapper采用分子形状叠加和化合物特征匹配相结合的内部SHAFTS方法进行三维相似度搜索、排序和叠加。以用户提供的化学结构作为查询,SHAFTS将数据库中的每个化合物与查询分子对齐并计算三维相似度得分,返回最相似的结构。在这些最相似的结构基础上,构造出一个化合物-蛋白质网络,并采用随机漫步算法计算查询结构与命中化合物相关的蛋白质之间相互作用的概率。最后将这些潜在的蛋白质靶点按概率的标准分数排名。只需绘制查询分子或上传分子文件,操作简单。
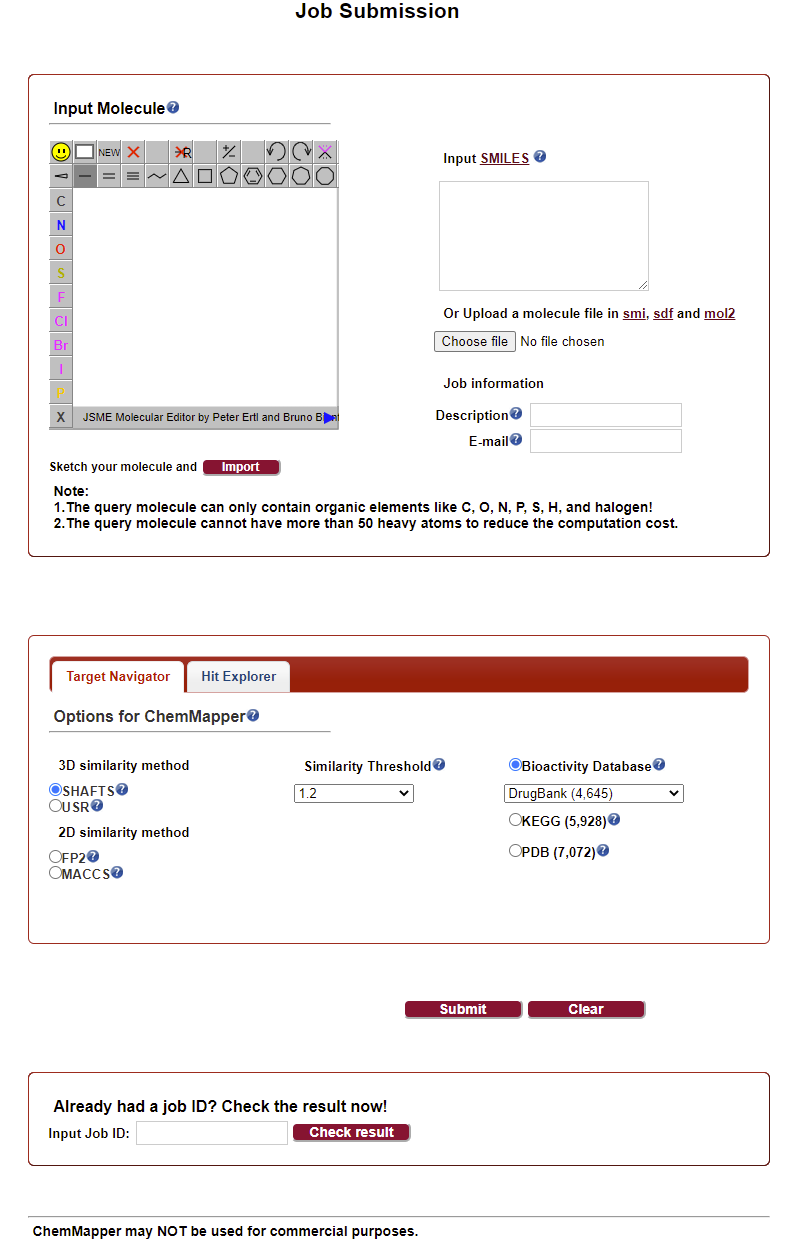
07 SEA
SEA工具是Michael团队开发的基于配体结构相似性预测靶点的在线工具,文章发表于Nature method。开发者将65000 个配体-靶标注释信息分成数百个药物靶点的集合,使用配体拓扑计算每组之间的相似性分数,开发了一个统计模型来对得到的相似性分数的重要性进行排序。该算法可用于大型复合数据库的快速搜索和跨靶标相似性图的构建。
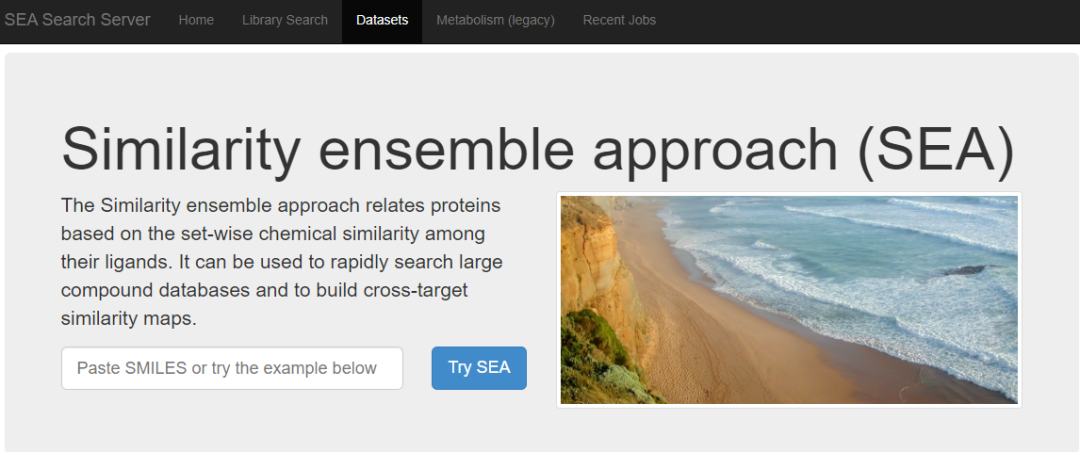
08 RF QSAR
识别靶标的著名计算机方法之一是结构活性关系,韩国科学技术院开发了一个基于配体的虚拟筛选模型,包括使用随机森林算法构建的 1121 个靶标 SAR 模型。通过使用 ROC 曲线和内部五折交叉验证的平均分数来测试每个靶标模型的性能。此外,计算前 k 个靶标的召回率以评估靶标排名的性能。通过外部验证集检查了使用优化采样方法和参数的基准模型。结果显示,前 11 名(占总靶标的 1%)和前 33 名的召回率分别为 67.6% 和 73.9%。
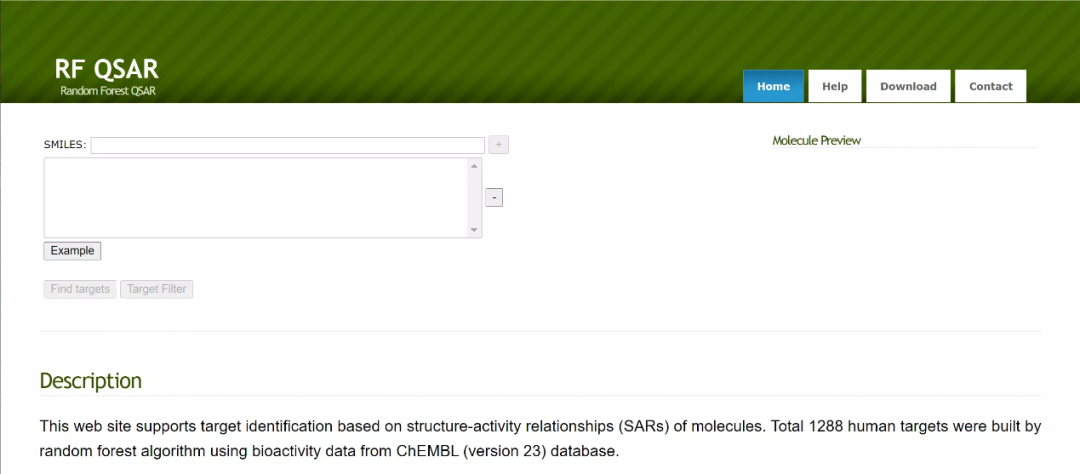
09 SwissTargetPrediction
SwissTargetPrediction基于与已知化合物的二维和三维结构的相似性来预测化合物的靶标。预测可以在人、大鼠、小鼠三种不同物种中进行。已知的化合物-靶标相互作用来自第16版的ChEMBL数据库,它由280381个小分子与2686个靶标间的相互作用构成,其中大多数靶标(66%)是人的蛋白。SwissTargetPrediction为每个预测靶标提供一个分数,以评估预测正确的可能性。它还通过不同物种之间的同源性映射进行预测,并提供正确可能性得分。2014年上线,界面友好,简单好用~
10 Anglerfish
Anglerfish也是一个基于配体相似性来预测药物靶标的在线服务器,提供查询分子结构文件后可自主选择相似性方法。
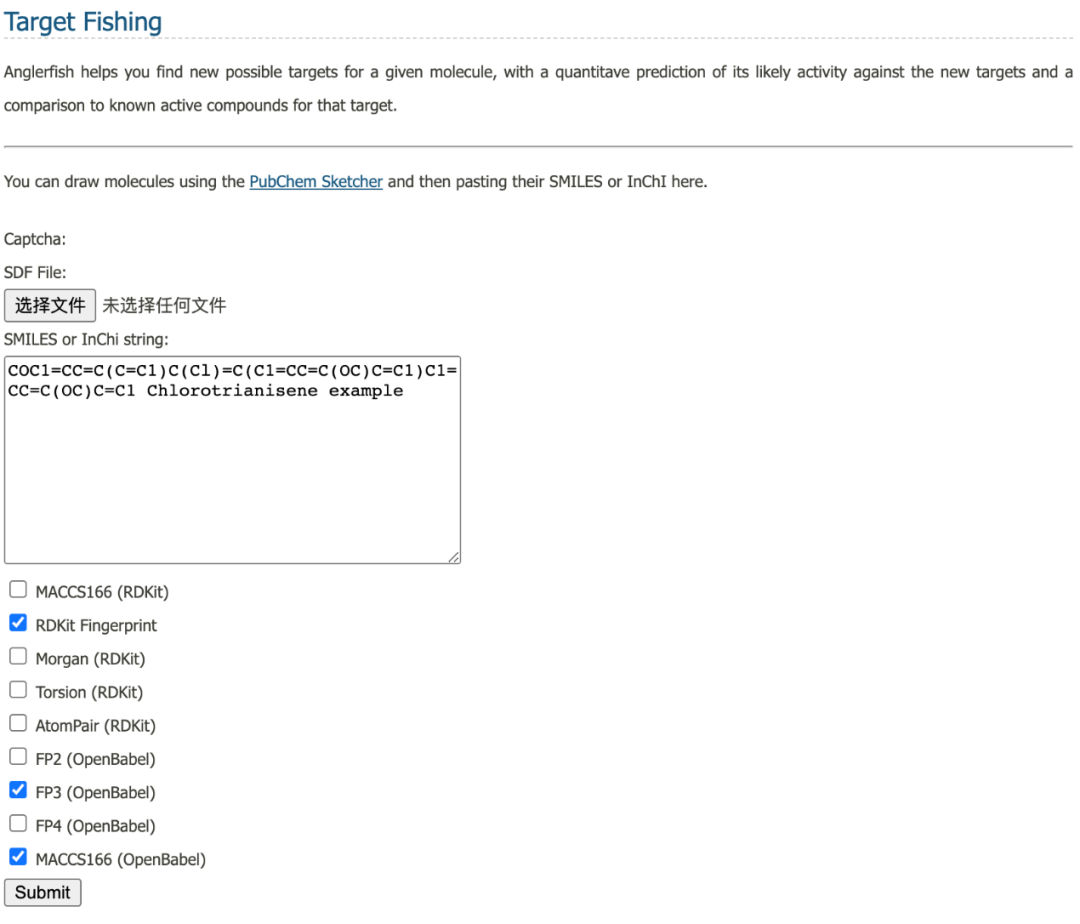
11 SuperPred
SuperPred是预测ATC编码和化合物靶标的在线服务器。ATC 预测和靶标预测基于机器学习模型,使用逻辑回归和长度为 1024 的摩根指纹。同时考虑了碎片的三维相似性、碎片的出现以及物理化学性质的一致性。
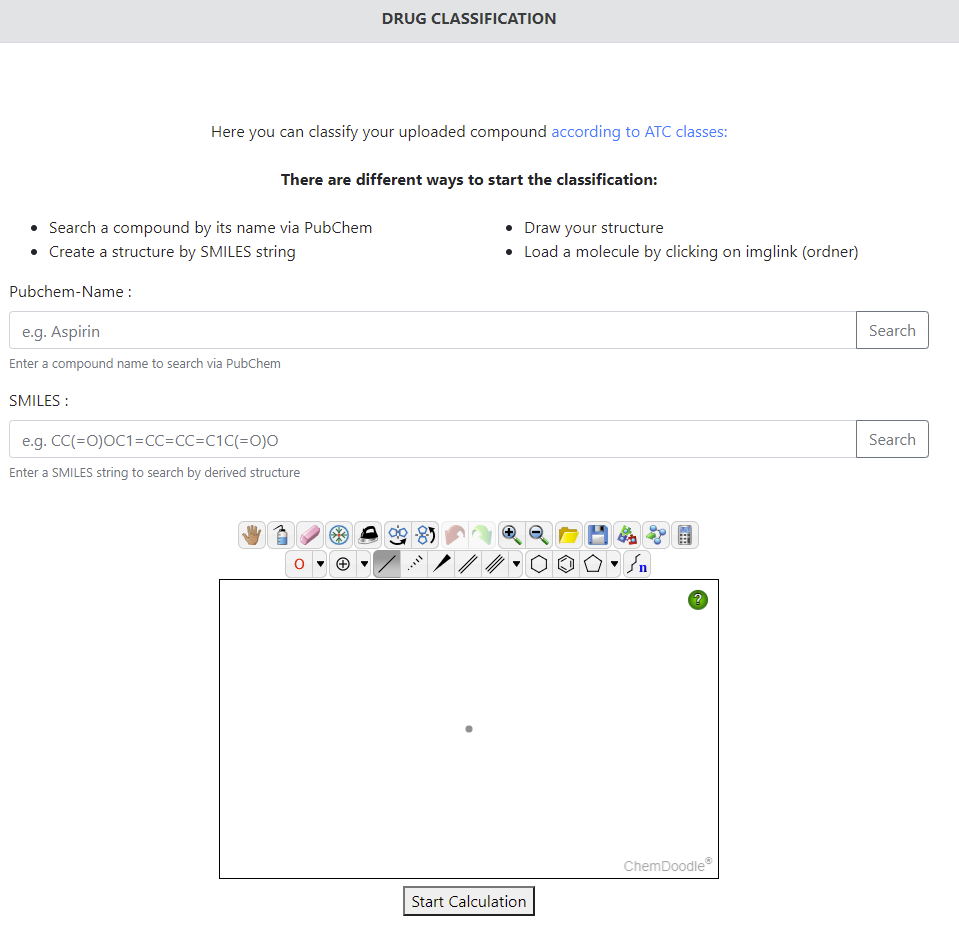
12 COVIDep
港科大电子及计算机工程学系兼化学及生物工程学系Matthew McKay教授带领的研究团队,近日设立了首个新冠病毒疫苗研发大数据平台“COVIDep”。平台采用全球基因数据库GISAID信息,为现时每日持续新增发现的新冠肺炎病毒基因序列,与对SARS 病毒呈免疫反映的B细胞和T细胞抗原表位进行相似度分析,从而筛选出有可能触发新型肺炎免疫反应的靶点,为正在研究开发疫苗的科学家提供重要指引。
13 idTarget
idTarget 是一个网络服务器,可以通过对接方法预测小化学分子的可能结合靶标,并结合最近开发的蛋白质靶标的评分函数和亲和力概况训练了新的稳健评分函数,函数基于稳健的回归分析和量子化学电荷模型。在分子对接计算中,构建小的重叠网格来约束搜索空间,从而更高效地实现对接结果的收敛。idTarget 已被证明能够重现已知的药物或药物样化合物的脱靶。

14 GalaxySagittarius
这是一个预测类药化合物潜在靶标的网站。原理是基于配体结构的相似性,以及预测的结合位点与可用配体信息的蛋白质靶标的相容性。通过使用蛋白质-配体对接程序 GalaxyDock2 将查询化合物与预测模式中的 1,000 种蛋白质中的每一种对接,以重新排列 预测靶标并获得靶标-化合物的复合结构。
15 MolTarPred
预测基于计算得到的查询分子的 Morgan 指纹与要筛选的 607,659 个 ChEMBL 化合物中的每一个之间的 tanimoto 相似性,预测靶标为从与查询分子最相似的前 10 个分子中检索到的靶标。结果页面包括预测的靶标信息、预测的置信度分数以及与预测靶标结合的命中结构相似性的可视化图。

















 1294
1294

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









