






 本文介绍了PLINK工具中常见的格式转换操作,包括ped/map到bed/bim/fam、tped/tfam等,以及如何进行SNP质量和性别检查,同时提供了使用Python脚本更新bim文件的示例。
本文介绍了PLINK工具中常见的格式转换操作,包括ped/map到bed/bim/fam、tped/tfam等,以及如何进行SNP质量和性别检查,同时提供了使用Python脚本更新bim文件的示例。
目录
ped/map -> bed/bim/fam、tped/tfa
bed/bim/fam -> ped/map、tped/tfam、vcf、bgen
tped/tfam -> bed/bim/fam、ped/map
vcf -> ped/map、bed/bim/fam、tped/tfam
格式转换
plink常见格式类型
- 一般格式:ped/map
- 二进制格式:bed/bim/fam (建议)
- 转置格式:tped/tfam
ped/map -> bed/bim/fam、tped/tfa
# ped/map -> bed/bim/fam
plink --file arab --make-bed --out arab
# ped/map -> tped/tfam
plink --file arab --recode --transpose --out arab bed/bim/fam -> ped/map、tped/tfam、vcf、bgen
# bed/bim/fam -> ped/map
plink --bfile arab --recode --out arab
# bed/bim/fam -> tped/tfam
plink --bfile arab --recode --transpose --out arab
# bed/bim/fam -> vcf
plink --bfile arab --export vcf --out arab
# bed/bim/fam -> bgen
plink2 --bfile arab --export bgen-1.2 --out arabtped/tfam -> bed/bim/fam、ped/map
# tped/tfam -> bed/bim/fam
plink --tfile arab --make-bed --out arab
# tped/tfam -> ped/map
plink --tfile arab --recode --out arab vcf -> ped/map、bed/bim/fam、tped/tfam
# vcf -> ped/map
plink --vcf arab.vcf --recode --out arab
# vcf -> bed/bim/fam
plink --vcf arab.vcf --make-bed --out arab
# vcf -> tped/tfam
plink --vcf arab.vcf --recode --transpose --out arab--file/bfile/tfile定义输入格式为ped/map、bed,bim,fam、bed,bim,fam--vcf 定义输入格式为vcf, 压缩(.vcf.gz)或者不压缩(.vcf)都可以接受--recode定义输出格式为ped/map--make-bed定义输出格式为bed,bim,fam--recode --transpose定义输出格式为tped/tfam--export 指定输出的格式
质量控制
# 第一次筛选
plink --file input --geno 0.05 --mind 0.1 --maf 0.05 --hwe 0.000001 --make-bed --out output
# 性别检测
plink --file output --check-sex # 生成plink.sexcheck
grep "PROBLEM" plink.sexcheck| awk '{print$1,$2}'> sex_discrepancy.txt
plink --file output --remove sex_discrepancy.txt --make-bed --out output2
# 再次筛选
plink --file output2 --geno 0.05 --mind 0.1 --maf 0.05 --hwe 0.000001 --make-bed --out output3--file/bfile/tfile定义输入格式为ped/map、bed,bim,fam、bed,bim,fam--geno指定SNP缺失阈值。如果一个SNP在超过5%的sample中都是缺失的,那么就删掉该SNP。--mind指定sample缺失阈值。如果一个sample有超过5%的SNP都是缺失的,那么就删掉该sample。--hwe 哈代平衡。根据基因型频率进行筛选。--check-sex 生成性别检测的结果,输出默认名为removesample.txt--remove 根据removesamp.txt将性别出错的样本剔除。--make-bed定义输出格式为bed,bim,fam。
删除特定SNP位点
plink --file input --exclude xxx.txt --make-bed --out output--file/bfile/tfile定义输入格式为ped/map、bed,bim,fam、bed,bim,fam--exclude 指定描述了需要删除的SNP的文件--make-bed定义输出格式为bed,bim,fam
xxx.txt为一列特定SNP identity的纯文本文件:
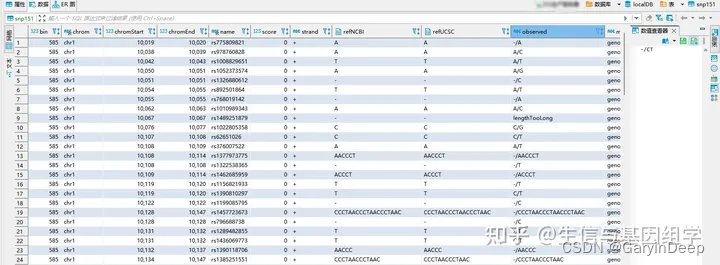
bim文件: rs编号 <-> SNP染色体/位置
1、从UCSC下载纯文本格式的dbSNP release 151 并解压。二选一hg19/hg38、snp151/snp151Common
#hg19 版本
#snp151.txt.gz 包含所有SNPs,共12G
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp151.txt.gz
gzip snp151.txt.gz -d
# snp151Common.txt.gz包含常见SNPs,共748M
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp151Common.txt.gz
gzip snp151Common.txt.gz -d#hg38 版本
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/database/snp151.txt.gz
gzip snp151.txt.gz -d
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/database/snp151Common.txt.gz
gzip snp151Common.txt.gz -d注意:下载的xxx.txt的 chromStart 列是 0-based 的。
二、编写 Python 脚本,脚本名字为 update.py:
import sys
snps_dic = {}
for item,row in enumerate(open(UCSC_path, encoding="utf-8")): # 载入字典 0-base
list = row.strip().split("\t")
snps_dic[list[4]] = list[2]
bim = open(sys.argv[2], encoding="utf-8")
new = open(sys.argv[3], 'w')
for row in bim:
list = row.strip().split("\t")
list[1] = snps.get(list[1]+1, list[1]) # 替换 0-base->1-base
new.write('\t'.join(list) + '\n')
new.close()
bim.close()运行脚本文件,original.bim为原本的bim文件, new.bim为更新后的bim文件。
python update.py snp151.txt original.bim new.bim由于snp151.txt内含有超过6亿个SNP信息,建议根据本模板重新编写脚本以实现分段读取snp151.txt。

















 2971
2971

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









