







PED/MAP/VCF文件与PLINK-BED/BIM/FAM文件格式相互转换
1. 主要文件格式简介
1.1 PED/MAP 格式
.ped文件:样本基因型数据
.map文件:SNP 位置信息
1.2 BED/BIM/FAM 格式
.bed文件:二进制基因型数据
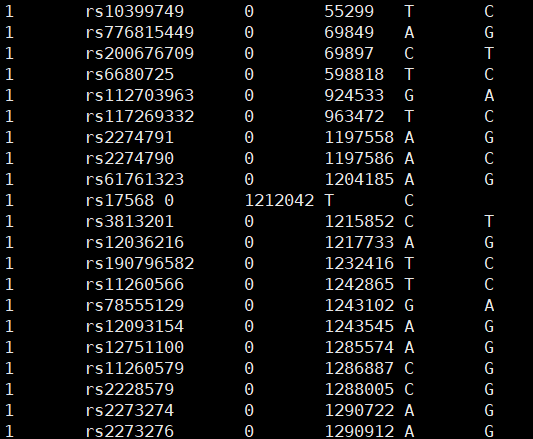
.bim文件:SNP 信息
.fam文件:样本信息
2. PLINK过滤参数
2.1 过滤低质量位点
(1)位点缺失率 (–geno): 过滤掉缺失率过高的位点,低深度数据中,位点缺失率较高,适当放宽
阈值以保留更多位点,建议值:–geno 0.1(过滤掉缺失率 >10% 的位点)。
(2)最小等位基因频率 (–maf):过滤掉低频变异位点,因为低频位点可能是测序错误,过滤后可
以提高数据质量,建议值:–maf 0.01(过滤掉最小等位基因频率 <1% 的位点)。
(3)哈迪-温伯格平衡 (–hwe):过滤掉偏离哈迪-温伯格平衡的位点,偏离平衡的位点可能是测序
错误或选择压力的结果,建议值:–hwe 1e-6(过滤掉显著偏离平衡的位点)。
2.2 过滤低质量样本
(1)个体缺失率 (–mind): 过滤掉缺失率过高的个体,低深度数据中,个体缺失率较高,适当放宽
阈值以保留更多样本,建议值:–mind 0.1(过滤掉缺失率 >10% 的个体)。
(2)性别一致性检查 (–check-sex): 检查样本性别与基因型数据的一致性,建议值:结合 –
check-ex 参数使用,手动检查并过滤不一致的样本。
2.3 其他过滤参数
(1)最小分型率 (–geno 和 --mind 结合): 综合过滤位点和个体的缺失率,建议值:根据数据质量
调整,例如 --geno 0.1 --mind 0.1。
(2)连锁不平衡过滤 (–indep-pairwise): 过滤掉高连锁不平衡的位点,适用于后续的 PCA 或关联
分析,建议值:–indep-pairwise 50 5 0.2(窗口大小 50 SNP,滑动步长 5 SNP,LD 阈值 0.2)。
3. VCF转换为PED+MAP
plink --vcf input.vcf --recode --out output
4. PED+MAP转换为BED+BIM+FAM
plink --file input --make-bed --out output
5. BED+BIM+FAM转换为PED+MAP
plink --bfile input --recode --out output
6. 23andMe 格式转换为 PED+MAP
plink --23file input.txt --recode --out output
7. VCF转换BED/BIM/FAM PLINK文件(无参数)
# output_prefix:输出文件前缀
# 压缩VCF
plink --vcf input.vcf.gz --make-bed --out output_prefix
# 未压缩VCF
plink --vcf input.vcf --make-bed --out output_prefix
8. VCF转换为BED/BIM/FAM PLINK文件(过滤参数)
生成output_prefix.fam, output_prefix.bim, output_prefix.bed 三个文件及分析日志log等文件。
plink --vcf input.vcf --make-bed --out output_prefix \
--geno 0.1 \ # 过滤缺失率 >10% 的位点
--mind 0.1 \ # 过滤缺失率 >10% 的个体
--maf 0.01 \ # 过滤最小等位基因频率 <1% 的位点
--hwe 1e-6 \ # 过滤偏离哈迪-温伯格平衡的位点
--indep-pairwise 50 5 0.2 # 过滤高连锁不平衡的位点
bim文件:



















 9757
9757

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











