






论文地址:http://zzdlab.com/upload/BIB-20230110-HuangYan.pdf
摘要:
本篇论文使用了深度学习网络预测蛋白质-蛋白质相互作用,网络主体是一个基于GCNN(图卷积神经网络)的暹罗结构用于提取蛋白质特征,后续是一个分类器,使用一个全连接作为分类器。本文最大的创新处在于SGPPI网络专注于蛋白质-蛋白质结合界面上的蛋白质斑块,并从残基接触图中提取结构、几何和进化特征来预测PPI,而非使用原有序列和化学性质等一维理化性质作为预测信息。
输入信息:
数据集:蛋白质相互作用相似性限制:为了限制蛋白质序列相似性的影响,通过将序列同一性阈值设置为40%来消除相互作用蛋白质的序列冗余。使用三个基准测试集Profppikernel dataset, Human Reference Interactome (HuRI) dataset and (filtered) Pan’s dataset作为基准测试集。最终阴性:阳性数据集=10:1。
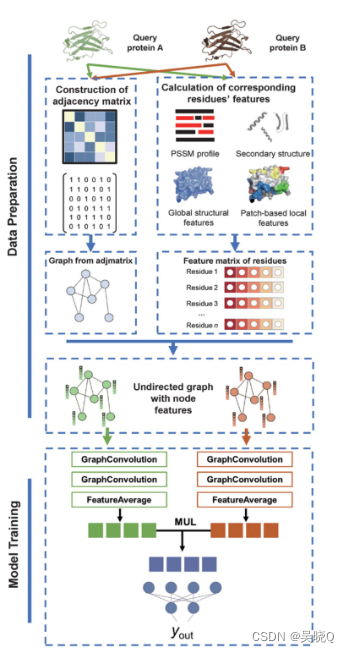
重点数据:蛋白质上面重要的有两个区域:Surface residues和Patch residues。Surface residues指的是蛋白质表面上的氨基酸残基,与溶剂相接触。而Patch residues是指蛋白质表面上一块区域内的氨基酸残基,这些残基通常具有特定的化学性质或功能。
输入数据1局部和全局几何特征(包含上述重点数据):此部分使用JET2特征编码提取。JET2编码是一种专用的蛋白质surface patch识别算法,它将每个蛋白质界面从核心部分到边缘部分划分为三个结构区域,称为种子、延伸和外层。使用保守性水平 ,残疾进化轨迹,界面倾向一级蛋白质结构计算的循环方差(CV)来表征其特征,其中CV分为局部CV和全局CV。
输入数据2 PSSM(编码进化信息的PSSM)使用PSSM图谱来重新选择每个氨基酸残基的保守性和突变图谱。
输入数据3 蛋白质二级结构信息,使用DSSP鉴定蛋白质的二级结构,将其编码成八维独热编码。DSSP (Dictionary of Secondary Structure of Proteins) 是一种用于分析蛋白质二级结构的计算机程序。它可以将蛋白质原子坐标信息输入并输出每个氨基酸残基的二级结构类型和氢键等详细信息
模型结构:
模型采用了一种图卷积神经网络,使用残差接触图形成图结构,定义图结构为G=(V,E),V表示所有的残基,E表示残基-残基接触。(接触:任何两个残基的Cα原子的几何距离小于某个阈值(默认值为10Å))。而氨基酸序列、蛋白质二级结构、几何特征和进化信息特征被编码并映射到每个节点上。经过图卷积和分类器得到预测结果。
结果:
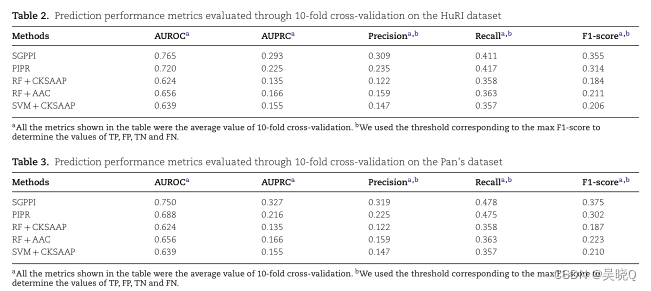
启发:
优点:之前看到很多图结构用于表示每个蛋白质互作而形成的图结构,每个节点性质为该蛋白质的性质,此论文使用图表示残差接触,每个节点放氨基酸的信息,是一种启发可以将三级空间结构加入。















 627
627











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









