






生信碱移
EGG 估计遗传图
EGG 是由美国凯斯西储大学的研究者开发的 R 包,其提出了一个用于理解性状间依赖关系的方法,该方法使用多变量孟德尔随机化中使用的相同技术来消除估计误差偏差和水平多效性偏差。具体来讲,通过利用全基因组关联研究(GWAS)数据,该方法首先校正数据中的估计误差和水平多效性偏差,然后采用熵损失函数和交替方向乘子法(ADMM)优化算法估计协方差矩阵和精度矩阵,并通过非凸惩罚函数选择重要的遗传效应,最终构建一个显示多种性状间关系的高斯图模型,从而有效地理解和可视化这些性状之间的生物学依赖关系。

虽然 EGG 目前仍然处于预印本阶段,但是其通讯作者今年还是刚刚发了一篇 NC 的方法开发,所以整体而言还是有一定可信度

。
github 链接:
-
https://github.com/harryyiheyang/EGG
1、EGG的安装
用户可以通过以下代码从gihub安装EGG:
# 安装 EGG
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("harryyiheyang/EGG")
# 其他所需的依赖包
install.packages(c("data.table", "glasso"))
2、示例数据加载
这里展示 EGG 内置示例数据的使用,如果大家用的是自己的数据,整理成相应的格式即可:
library(igraph)
library(RColorBrewer)
library(EGG)
data("EAS_Error_COV")
data("EAS_Zscore")
BETA <- EASZZ[,-1]
R <- EASZR
简单看看所需的两个输入数据BETA和R:
# BETA 表头
BETA[1:5, 1:5]
CAD BMI TBil PLA RBC
rs2210366 1.870127 0.1396186 0.7485899 -24.775181 34.801015
rs295 -4.195356 0.1945283 -2.3653577 3.004525 -1.135550
rs11216126 -3.554459 2.7019135 -1.2092108 4.358674 1.465796
rs11065774 15.205588 -3.9030912 -6.8720610 10.585175 9.926085
rs3782886 18.777007 -6.1186400 -8.6412653 15.702205 14.281071
# R 表头
R[1:5, 1:5]
CAD BMI TBil PLA RBC
CAD 0.893202619 0.029882288 -0.009896194 -0.00207113 -0.001764895
BMI 0.029882288 1.023218706 -0.002950979 0.01326042 0.124089753
TBil -0.009896194 -0.002950979 0.882567949 -0.12085292 0.114197283
PLA -0.002071130 0.013260415 -0.120852922 0.93052917 0.020903933
RBC -0.001764895 0.124089753 0.114197283 0.02090393 0.922086721
简单看看文档里面对这两个输入文件的介绍:
-
「BETA」: A matrix of dimensions mxp, where m is the number of variants and p is the number of exposures. It represents effect size estimates or Z-scores.
-
「Rnoise」:The covariance matrix of estimation errors in BETA. If BETA contains Z-scores, Rnoise should be a correlation matrix.
比较明确,BETA 是 GWAS 结果中的效应值beta或者Z-score矩阵,列对应性状,行对应 snp,填充值对应beta或z-score。Rnoise 是 GWAS 结果中效应值对应标准误的协方差矩阵。多插一嘴,误差的协方差是两个变量之间共同变化的度量,如果两个变量的误差倾向于同时增加或减少,则它们具有正协方差;如果一个增加而另一个减少,则它们具有负协方差。在 GWAS 中,不同性状之间的效应值误差可能会有类似的协方差,这反映了它们的共同关联。
3、执行分析并可视化
# 执行分析, 具体的参数大家可以看看
fit1 <- entropy.mcp.spearman.sampling(
BETA, # BETA 矩阵,维度为 m x p,其中 m 是变异数,p 是性状数,表示效应大小估计或 Z 分数
Rnoise = R, # 估计误差的协方差矩阵,如果 BETA 包含 Z 分数,Rnoise 应为相关矩阵
lamvec = c(25:52)/1125, # MCP(最小凹惩罚)的调优参数向量,范围为 25 到 52 除以 1125
max.eps = 0.005, # 算法停止的阈值,当算法的变化小于此值时停止,默认值为 0.01
max.iter = 25, # 算法的最大迭代次数,ADMM 收敛快但精度不高,因此不宜过大,默认值为 25
rho = 0.05, # ADMM 算法的惩罚参数,默认值为 0.05
mineig = 0.01, # 精度矩阵估计的最小矩阵特征值,默认值为 0.01
subfrac = 0.5, # 每次重新采样的数据比例,默认值为 0.5
subthres = 0.5, # 稳定性选择的阈值,默认值为 0.95
subtime = 300, # 重新采样的次数,默认值为 100
alpha = 0 # MCP + alpha * Ridge 的附加参数,默认值为 0
)
# 看看交叉验证的损失变化
plot(rowMeans(fit1$cv.error), main = "CV Error of EGG")
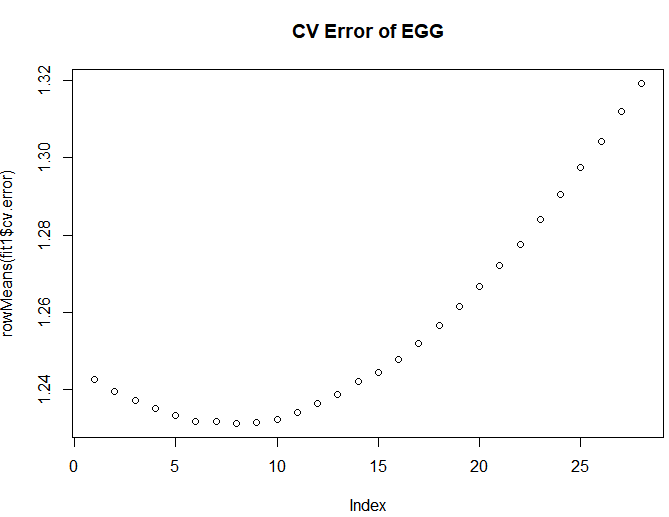
▲ 交叉验证的损失变化
可视化一下:
ThetaEUR <- -fit1$Theta * (fit1$K > 0.95)
KEUR <- fit1$K
nam <- rownames(ThetaEUR)
colnames(ThetaEUR) = rownames(ThetaEUR) = nam
nam <- sort(nam)
ThetaEUR <- ThetaEUR[nam, nam]
gEUR <- graph_from_adjacency_matrix(ThetaEUR, weighted = TRUE, diag = FALSE, mode = "undirected")
V(gEUR)$size <- 12
E(gEUR)$color <- ifelse(E(gEUR)$weight > 0, "#ff7676", "#75B7D1")
pdf(file = "network.pdf", width = 10, height = 10)
plot(gEUR, layout = layout.circle,
vertex.size = V(gEUR)$size+5,
vertex.label = V(gEUR)$name,
vertex.label.cex = 2,
vertex.label.color = "black",
edge.color = E(gEUR)$color,
vertex.frame.color = V(gEUR)$color,
edge.width = 2, main = "Network estimate of EGG")
dev.off()
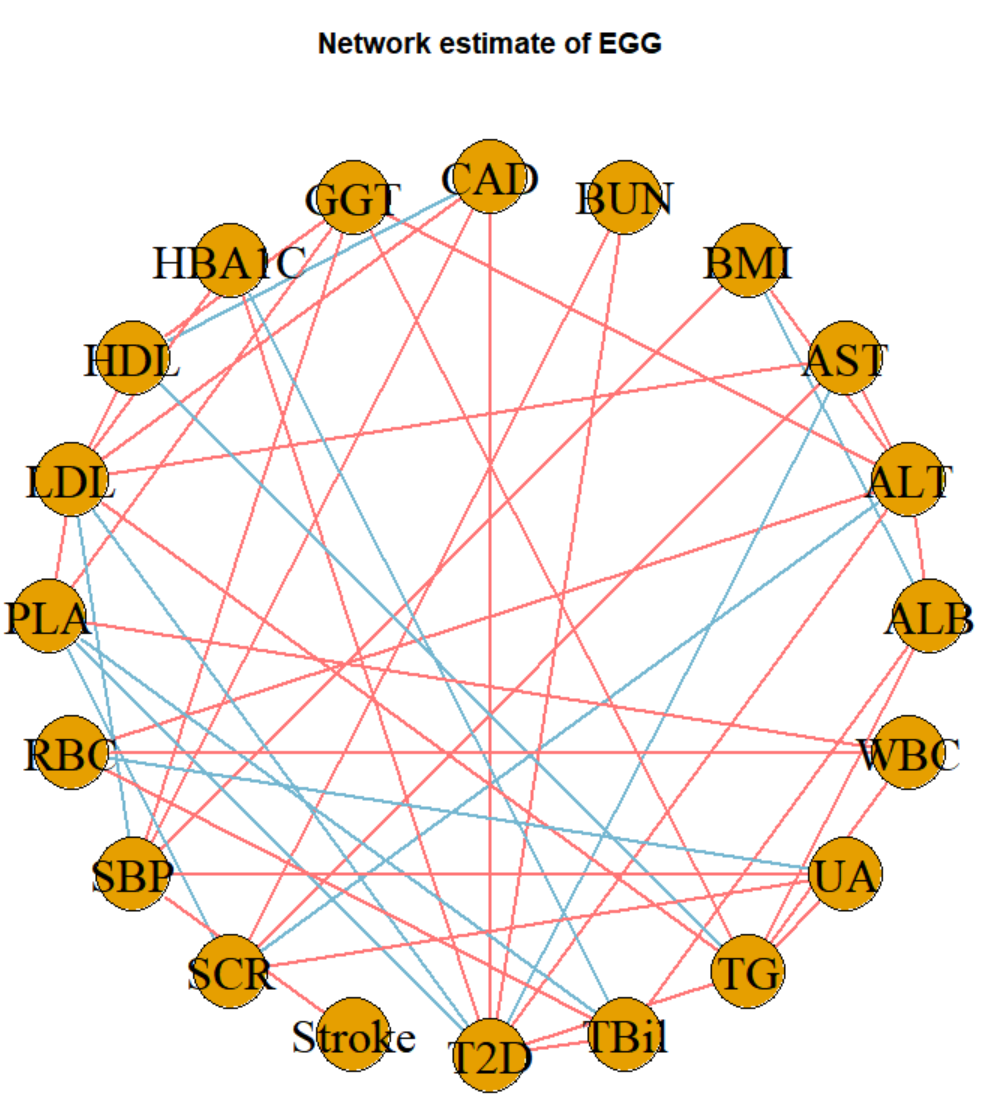
▲ EGG 方法的可视化结果:由于上方代码的设置,蓝色的边表示性状之间存在负条件相关性,红色的边表示正条件相关性。举个例子,在包括的其他性状的条件下,BMI 与收缩压 SBP 存在正相关。
简单分享到这















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









