






生信碱移
sclTD: 细胞状态协变算法
单细胞分析中,来自同一细胞类型的基因表达在不同个体之间也是存在差异的,这种差异是由遗传和环境的结合驱动的。一个十分有趣的现象是,导致器官水平生理变化的疾病或环境刺激可能会改变多种细胞类型的基因协调表达。举个例子,先天免疫反应可能同时增加髓系细胞中趋化因子基因和内皮细胞中粘附分子基因的表达,因为这些两种细胞类型发挥协调作用,最后的以招募中性粒细胞到感染部位。换句话说,髓系细胞"打水"、内皮细胞"煮饭",大家最后合作吃到了饭(招募中性粒细胞到感染部位)。所以说在这种情况下,会存在跨细胞的、有规律的基因协调表达模式(髓系细胞上调趋化因子基因、内皮细胞上调粘附分子基因)。但是,目前还是缺少比较明确的算法工具来鉴定出不同的跨细胞的基因协调表达模式。

▲ 组织驻留的巨噬细胞可以对各种刺激(如细菌)作出反应,并分泌因子(细胞因子、趋化因子和生长因子),导致/协调内皮细胞的激活。血液中待被募集的细胞识别粘附分子,开始在内皮细胞层上滚动和粘附,随后穿过内皮细胞形成的紧密连接进入组织。图片来源:10.1007/s00424-017-1946-6。
在一个多维度测序数据中,塑造个体之间表达差异的潜在因素(例如,疾病亚型,后面也被称为因子)通常是未知的。相反,以无监督(无标签)方式分层个体的主要变异轴往往更具信息性。常见的矩阵分解方法,如主成分分析(PCA),可以应用于个体级别的基因表达数据,以提取任何个体细胞类型的轴。然而,使用这种分解来识别细胞之间的协调变异可能具有挑战性,因为来自不同细胞类型的主成分(PCs)通常显示出适度的、非唯一的关联。也就是说,在考虑个体的变异性时将不同的细胞亚群结合起来可以获得更多的信息,而不是单独只看一个细胞亚群或者仅将所有细胞亚群混为一体。
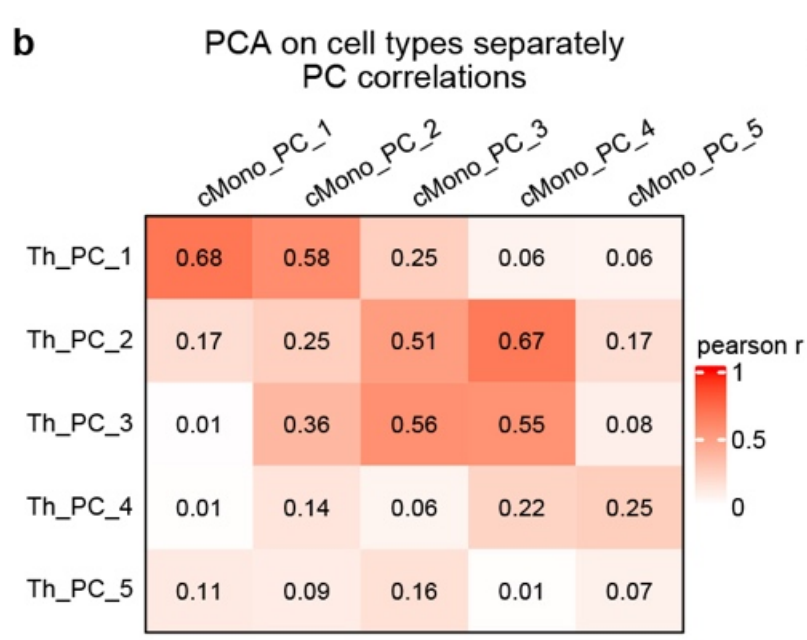
▲ 不同细胞类型的主成分(PCs)通常显示出一定程度上的、非唯一的关联。
为此,来自哈佛医学院与Altos实验室的研究人员开发了一种用于分析个体间变异的无监督方法,称为单细胞可解释张量分解scITD,于2024年9月23日发表于Nature Biotechnology [IF: 33.1]。

▲ DOI: 10.1038/s41587-024-02411-z
简单来讲,scITD针对每种细胞类型生成“供体×基因”的伪批量表达矩阵(这里的供体就是个体)。①这些矩阵通过细胞类型维度堆叠,形成一个三维张量(供体×基因×细胞类型)。②该张量在进一步分析之前会经过标准化、归一化和缩放等预处理步骤。③随后,作者使用Tucker张量分解将三维数据分解为几个因子矩阵(样本因子、基因因子、细胞类型因子)和一个核心张量。Tucker张量分解类似于PCA,但它是针对三维数据的。张量分解的结果包括:
-
样本因子矩阵(类似PCA中的样本得分):描述了供体/样本在潜在因子上的得分。
-
基因因子矩阵(类似PCA中的特征loading):描述了基因如何在这些潜在因子中表现。
-
细胞类型因子矩阵:描述了不同细胞类型之间的基因表达差异。
-
核心张量:这个张量反映了样本、基因和细胞类型之间的交互作用,类似于PCA中的协方差矩阵,但在三维空间上起作用。
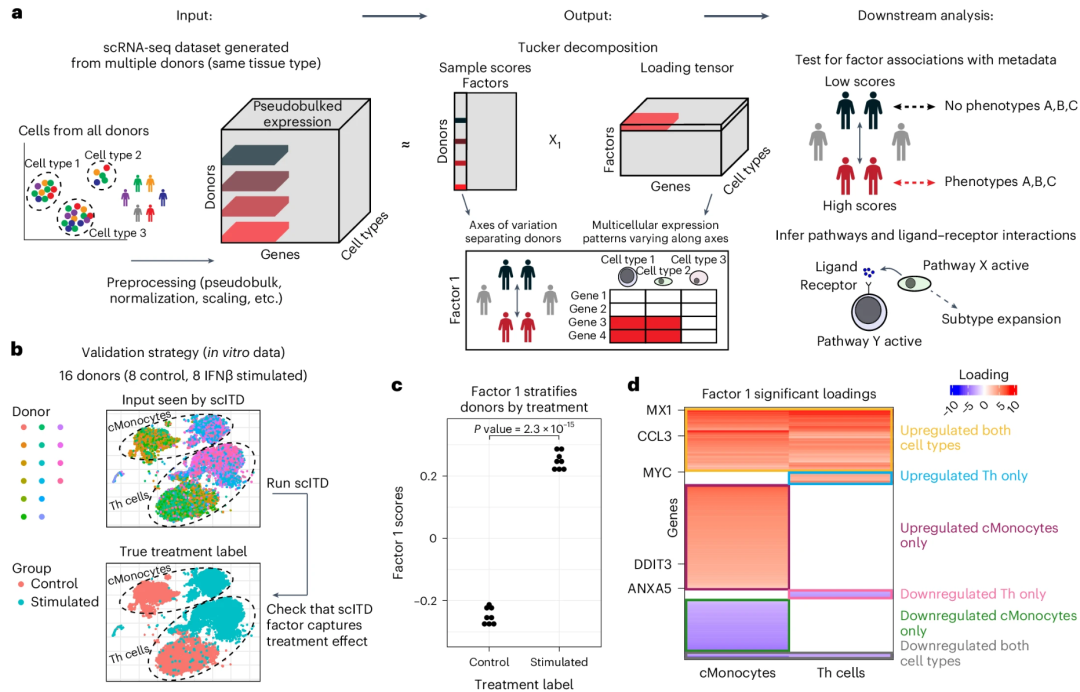
▲ scITD 的设计概述及功能演示。a, scITD 管道。左侧,输入包括一个 scRNA-seq UMI 计数矩阵和一个带有细胞类型和供体注释的元数据矩阵。中间,scITD 应用 Tucker 张量分解来识别由多种细胞类型的表达协变特征的个体间变异轴。右侧,得到的供体得分向量和多细胞模式可以进一步分析以寻找元数据关联和生物过程的特征。b, 使用 IFNβ刺激数据的验证策略,显示按供体或条件着色的 t 分布随机邻居嵌入图。c, 验证数据集中因子 1 的供体得分。供体按其真实治疗标签分组。使用线性模型F检验计算关联的显著性。d, 因子1的loading热图。仅显示在任一细胞类型中与因子 1 得分显著相关的基因loading(见原文方法学部分)。框表示在多种细胞类型组合中发现的上调或下调基因组。方向性表示处理供体相对于对照组的表达。
通过因子得分,用户可以将样本分为不同的表型组(如PhenotypesA,B,C),区分出具有相似基因表达模式的样本(上图中红色和黑色人形图标)。这里的因子,一部分可能便是本文开头提到的跨细胞的基因协调表达模式。除此之外,scITD支持的下游分析还包括推测基因调控通路、配体-受体相互作用及细胞亚型的扩展(如上图中提到的Pathway X、Pathway Y)。
本文只是介绍其主要运行代码,更多运行示例可以点击以下几个链接查看(本文示例数据来自下方第一个链接):
-
https://pklab.med.harvard.edu/jonathan/
-
https://pklab.med.harvard.edu/jonathan/LR_analysis.html
-
https://github.com/kharchenkolab/scITD
0.安装该包
运行以下代码安装该包,具体请查看下方代码注释:
#该软件包有几个来自 Bioconductor 的依赖包,要安装这些:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("ComplexHeatmap", "edgeR", "sva", "Biobase"))
# 从 GitHub 安装最新版本的 scITD
devtools::install_github("kharchenkolab/scITD")
01.使用指南
示例数据可以在下方链接头部中下载,其是来自van der Wijst等人(2018)的一项研究的45名健康供体外周血单核细胞的单细胞RNA-seq数据集:
-
https://pklab.med.harvard.edu/jonathan/
具体来讲,sclTD的输入包括:
-
行基因 、列细胞的UMI表达矩阵
-
每个细胞对应的元数据,对应seurat对象的meta.data
-
元数据必须包括标记为donors和ctypes的列,但它可以包括其他感兴趣的变量
①加载示例数据:
library(scITD)
#> Loading required package: Matrix
# counts matrix
pbmc_counts <- readRDS('/home/jmitchel/data/van_der_wijst/pbmc_counts_v2.rds')
# meta data matrix
pbmc_meta <- readRDS('/home/jmitchel/data/van_der_wijst/pbmc_meta_v2.rds')
# ensembl to gene name conversions
feature.names <- readRDS('/home/jmitchel/data/van_der_wijst/genes.rds')
②设置分析参数并创建分析项目:
# set up project parameters
param_list <- initialize_params(ctypes_use = c("CD4+ T", "CD8+ T", "cMonocyte", "CD56(dim) NK", "B"),
ncores = 30, rand_seed = 10)
# create project container
pbmc_container <- make_new_container(count_data=pbmc_counts,
meta_data=pbmc_meta,
gn_convert = feature.names,
params=param_list,
label_donor_sex = TRUE)
由于作者提供的元数据中没有关于供体/样本性别的信息,所以设置了label_donor_sex = TRUE以从性别特异性基因的表达中判断性别信息。这些新信息会自动添加到元数据中。虽然拥有这些信息并不是绝对必要的,但在后续分析中,如果识别出与性别相关的因子,这些信息可能会很有用。
③将数据形成一个维度为捐赠者X基因X细胞类型的张量。参数norm_method和scale_factor用于对伪合并数据进行归一化。参数vargenes_method和vargenes_thresh用于识别过度分散的基因。最后,scale_var和var_scale_power用于缩放最终张量中每个细胞类型切片中每个基因的方差。
# form the tensor from the data
pbmc_container <- form_tensor(pbmc_container, donor_min_cells=5,
norm_method='trim', scale_factor=10000,
vargenes_method='norm_var_pvals', vargenes_thresh=.1,
scale_var = TRUE, var_scale_power = 2)
#> parsing data matrix by cell/tissue type...
#> cleaning data...
#> Keeping 41 donors. All donors have at least 5 cells in each cell type included.
#> collapsing count matrices from cells to donors (aka pseudobulk operation)...
#> normalizing data...
#> calculating gene overdispersion factors...
#> selecting highly variable genes from each cell type...
#> scaling variance...
#> forming tensor...
#> Complete!
#运行上述函数后,应检查识别出的过度离散基因的数量
#以确保张量中有足够数量的基因,然后再进行分解
# number of genes included in the tensor
print(length(pbmc_container[["all_vargenes"]]))
#> [1] 741
#要增加包含的基因数量,只需使用更高的 vargenes_thresh 值重新运行 form_tensor()
④现在准备运行Tucker分解,并分析数据集中存在的有趣生物变异。指定rotation_type='hybrid'以表明使用混合旋转方法来优化因子载荷。这通过使基因模式更加模块化/相互独立来提高因子的可解释性。通过设置ranks=c(5,10),指定提取5个在供体/样本之间变化的多细胞过程(factor,因子),使用10个基因集。为了确定适当的设置,作者还在前面提到的教程链接的底部提供了一些有用的建议。
# run the tensor decomposition
pbmc_container <- run_tucker_ica(pbmc_container, ranks=c(5,10),
tucker_type = 'regular', rotation_type = 'hybrid')
⑤接下来,绘制供体/样本得分矩阵,该矩阵可以用于评估每个过程/因子(factor)在每个样本中的存在程度(不同的factor可以类比主成分,但是主成分的得分是每个样本才具有的,不是每个细胞都有)。同时还展示了得分与两个元数据变量sex和lanes(通道)之间的一些关联。
# get donor scores-metadata associations
pbmc_container <- get_meta_associations(pbmc_container, vars_test=c('sex','lanes'), stat_use='pval')
# plot donor scores
pbmc_container <- plot_donor_matrix(pbmc_container, meta_vars=c('sex','lanes'),
show_donor_ids = TRUE,
add_meta_associations='pval')
# show the donor scores heatmap
pbmc_container$plots$donor_matrix
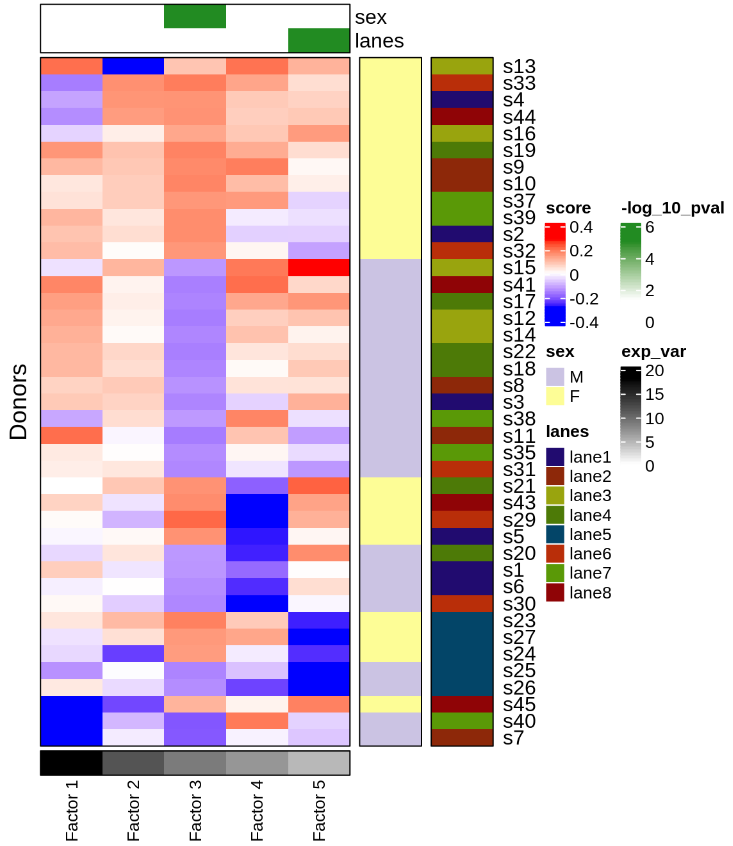
▲ 不同factor在每个样本中的得分,以及其余性状的潜在关联。
从上图中得分的变化不难看出,因子3与样本性别显著相关,因子5与10通道批次显著相关。
⑥确定每种细胞类型中与每个因子显著相关的基因。可视化每个因子的对应载荷矩阵,仅保留显著基因。这里的载荷矩阵其实可以类推PCA的载荷矩阵,Tucker分解对应的载荷只是加上了细胞类型的维度。也就是说,对于每种细胞类型,factor内部都有相应的基因得分。所以说,sclTD找到的factor对应本文开头所提及的跨细胞的基因协调表达模式(细胞亚群A具有更高的基因集m载荷,细胞亚群B具有更高的基因集n载荷,而基因集m、n被归类为一个factor,每个样本具有不同factor级别的评分,结合开头好好理解一下)。
# get significant genes
pbmc_container <- get_lm_pvals(pbmc_container)
# generate the loadings plots
pbmc_container <- get_all_lds_factor_plots(pbmc_container,
use_sig_only=TRUE,
nonsig_to_zero=TRUE,
sig_thresh=.02,
display_genes=FALSE,
gene_callouts = TRUE,
callout_n_gene_per_ctype=3,
show_var_explained = TRUE)
# arrange the plots into a figure and show the figure
myfig <- render_multi_plots(pbmc_container,data_type='loadings')
myfig
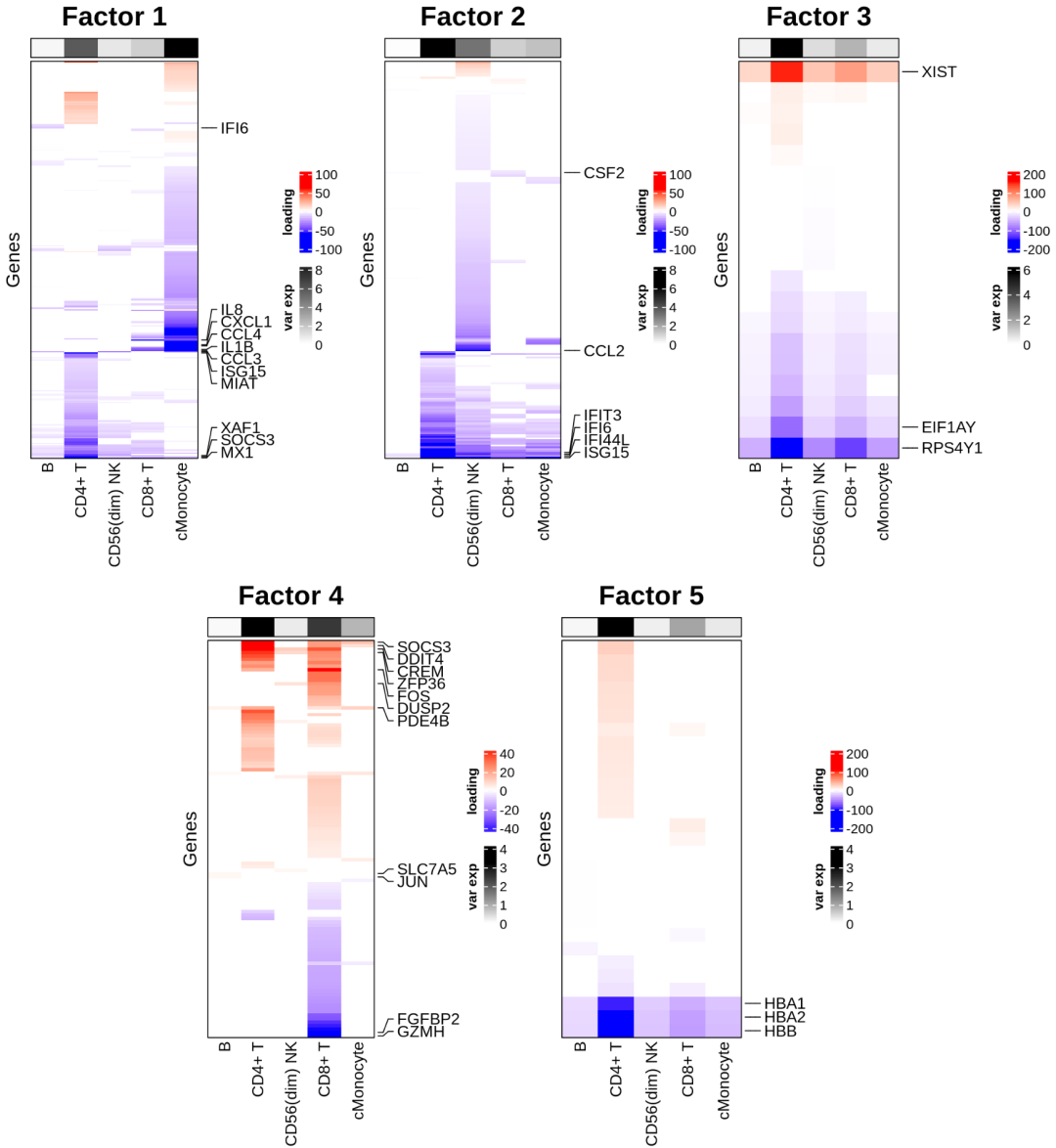
▲每种factor中显著基因在不同细胞中的载荷(loading)。
如上图所示看到,前面分析得出的与性别相关的因子(因子 3)的显著载荷包括X和Y染色体上的基因,这与预期一致。可以将正载荷基因(例如XIST)解释为在因子3(即女性)得分为正的供体(样本)中上调,反之亦然。同时还注意到,前面分析得出的与10通道相关的因子(因子5)涉及血红蛋白基因,这些基因在通道5中似乎上调,提示该通道批次可能存在一些红血球的污染。其他因子显示出一些有趣的模式,包含特定于不同细胞类型的过程。
⑦可以通过运行GSEA更好地了解与某个因子相关的细胞类型功能变化。以下函数可以使用多种功能注释集运行。在这里,使用factor_select参数指定因子1运行GSEA:
pbmc_container <- run_gsea_one_factor(pbmc_container, factor_select=1, method="fgsea", thresh=0.05, db_use=c("GO"), signed=TRUE)
#> Warning in preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (0.14% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> Warning in preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (0.41% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> Warning in preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (1.08% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
#> Warning in preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (3.25% of the list).
#> The order of those tied genes will be arbitrary, which may produce unexpected results.
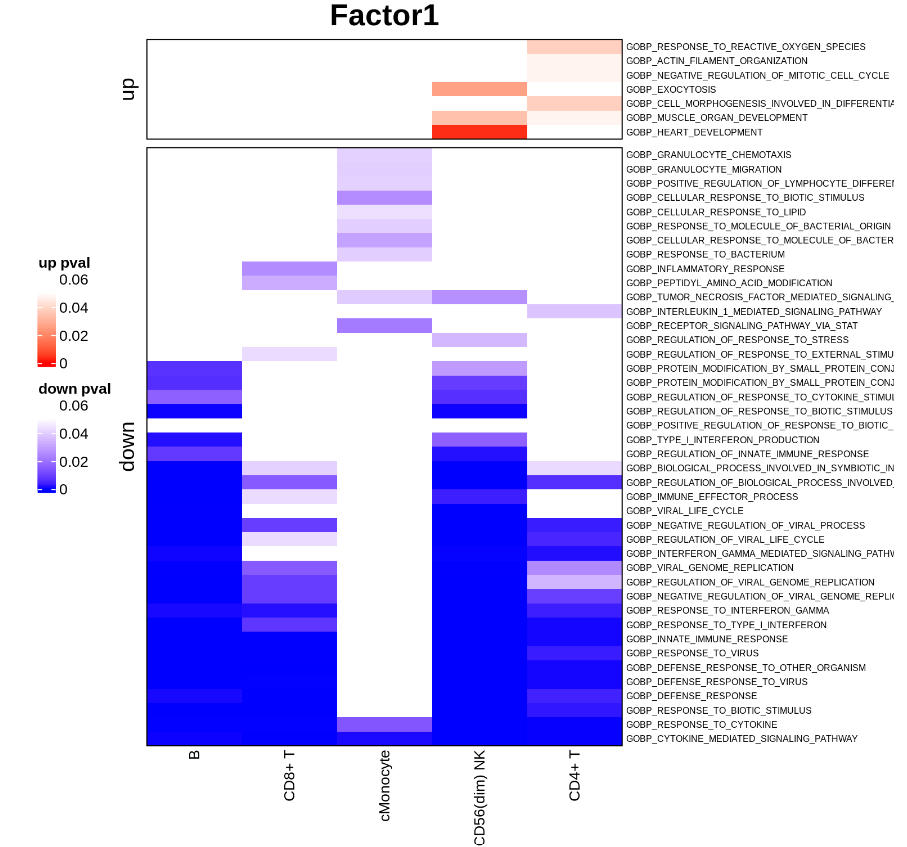
▲ factor1在不同细胞中的功能富集。
可以看到,因子1的特征是大多数细胞类型中干扰素刺激基因的富集,而单核细胞中则共同富集了与趋化相关的基因。将这一点与样本的评分联系起来,似乎这一多细胞过程(即facotr)整体上在样本s45、s40 和s7中最强烈上调。
选择最优的因子数量
如上小节的④中所述,在塔克分解中设置ranks参数还是比较模糊的。重申一下,该参数的第一个元素表示将要提取的因子的总数。参数的第二个元素用于内部,它大致表示共同变化基因的唯一集合数量。为了帮助选择适当的秩数量,作者实施了一种与矩阵分解中常用的策略类似的方法。简而言之,观察在增加因子数量时重构误差的变化,并将其与随机打乱数据集时观察到的误差变化进行比较。在这里,使用 SVD 计算沿捐赠者维度或基因维度展开的张量,以确定ranks参数的第一个和第二个元素的近似值。
# get assistance with rank determination
pbmc_container <- determine_ranks_tucker(pbmc_container, max_ranks_test=c(10,15),
shuffle_level='cells',
num_iter=10,
norm_method='trim',
scale_factor=10000,
scale_var=TRUE,
var_scale_power=2)
#> Warning: replacing previous import 'lifecycle::last_warnings' by 'rlang::last_warnings' when loading 'hms'
pbmc_container$plots$rank_determination_plot
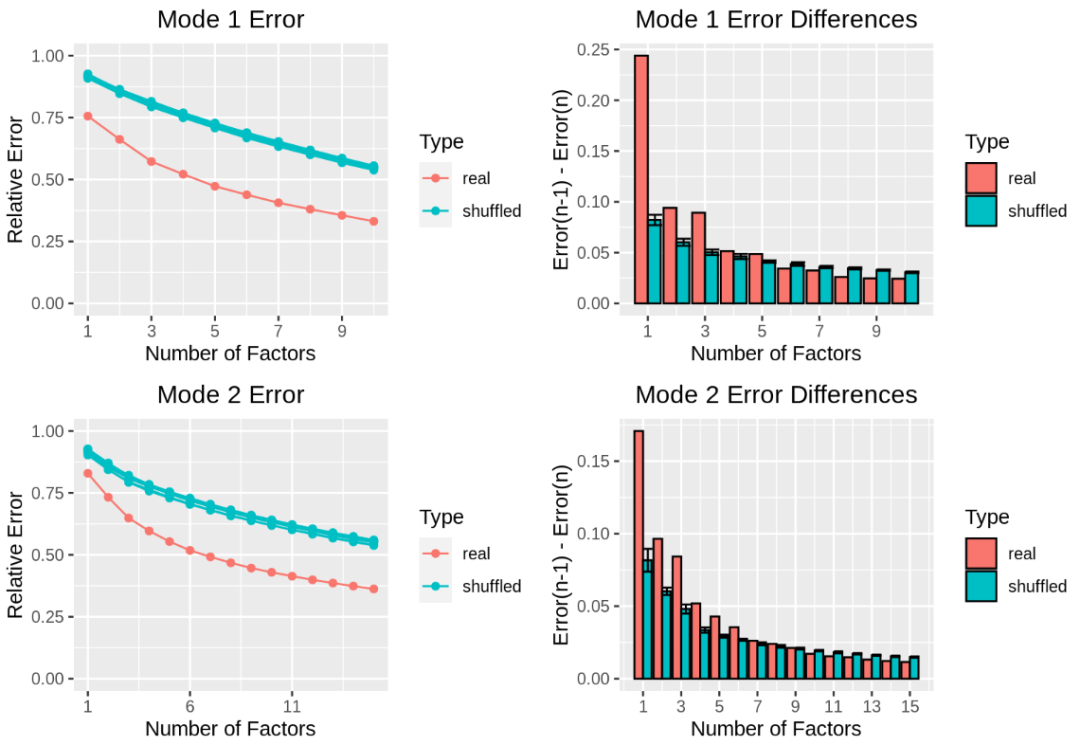
从右上角的图中可以看到,当因子的数量超过5时,误差的减少不再超过随机数据中所看到的误差。这就是为什么在上述分析中使用了5个主要因子的原因。右下角的图表建议使用大8个基因集合。
另一种帮助确定适当秩数的方法是评估在对数据集进行降解时,因子在子样本中保持的稳定性。如果提取了过多的因子,从而捕捉到噪声,那么这些因子可能不会表现出高重现性。为了应用这种方法,使用run_stability_analysis()函数,sub_prop参数确定在每次子样本迭代中保留的捐赠者的比例。
run stability analysis
pbmc_container <- run_stability_analysis(pbmc_container,ranks=c(5,10),n_iterations=50,subset_type='subset', sub_prop=.95)
pbmc_container$plots$stability_plot_dsc
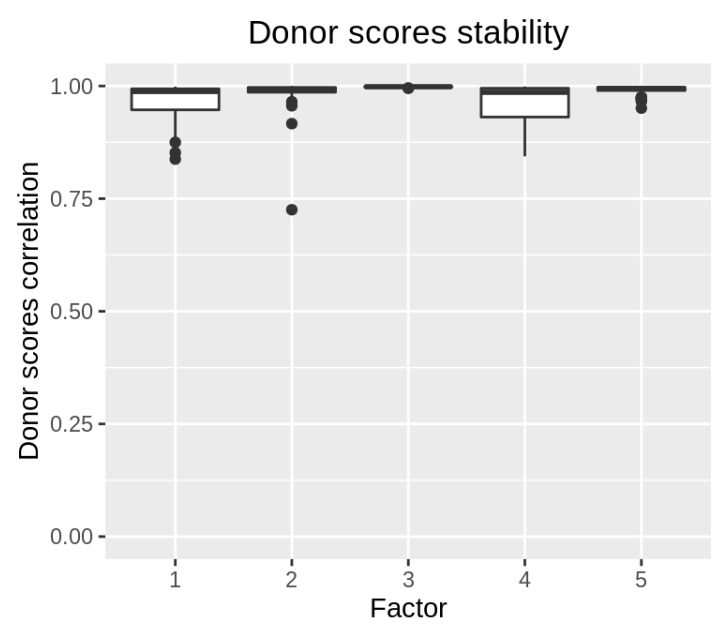
如上所示,可以观察到不同因子的良好稳定性,表明每个因子更可能代表生物学而非随机噪声。
今天就分享这
欢迎各位老哥老姐关注

















 378
378

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









