






第1章 原子结构
原子模型
- 道尔顿实心球模型:原子不可再分
- 汤姆逊枣糕模型:阴极射线
- 卢瑟福含核模型: α \alpha α 粒子轰击金箔
- 波尔原子轨道模型:光电效应
- 现代原子模型
波函数 ψ ( x , y , z ) \psi(x,y,z) ψ(x,y,z)
球坐标化的波函数
ψ nlm ( r , θ , φ ) = R nl ( r ) × Y lm ( θ , φ ) \psi_{\text{nlm}}(r,\theta,\varphi)=R_{\text{nl}}(r) \times Y_{\text{lm}}(\theta,\varphi) ψnlm(r,θ,φ)=Rnl(r)×Ylm(θ,φ)
波函数的物理意义
- ψ \psi ψ 对应核外电子运动的一种状态(原子轨道),对应不同的能量
- ψ 2 \psi^2 ψ2 为电子在某区域出现的概率密度
电子云
电子云:电子在原子核周围的概率分布,即 ψ 2 \psi^2 ψ2
径向分布函数
单位厚度球壳内电子分布概率
D
(
r
)
=
r
2
R
2
(
r
)
D(r)=r^2 R^2(r)
D(r)=r2R2(r)
量子数
| 量子数 | 对应 | 物理含义 |
|---|---|---|
| 主量子数 n = 1 , 2 , 3 , 4 , 5 , 6 , 7 n=1,2,3,4,5,6,7 n=1,2,3,4,5,6,7 | 能层 k , l , m , n , … \rm k,l,m,n,\dots k,l,m,n,… | 能量的主要影响因素,电子离核距离 |
| 角量子数 l = 0 , 1 , 2 , 3 ≤ n − 1 l=0,1,2,3\le n-1 l=0,1,2,3≤n−1 | 能级 s , p , d , f \rm s, p, d, f s,p,d,f | 电子角动量,电子云形状,多电子体系中对轨道能量有影响 |
| 磁量子数 m = 0 , ± 1 , ± 2 , ± 3 ≤ ± l m=0,\pm1,\pm2,\pm3 \le \pm l m=0,±1,±2,±3≤±l | 轨道 p x , p y , p z , … \rm p_x,p_y,p_z,\dots px,py,pz,… | 外加磁场时对电子能量略有影响 |
| 自旋量子数 m s = ± 1 / 2 m_{\text s}=\pm 1/2 ms=±1/2 | 电子自旋方向 | 电子本身的属性 |
原子轨道能量
单电子体系:轨道能量只与主量子数有关
E
n
=
−
Z
2
R
H
n
2
E_n=-\frac{Z^ 2R_H}{n^2}
En=−n2Z2RH
多电子体系(屏蔽与钻穿,
σ
\sigma
σ屏蔽系数)
{
E
n
=
−
Z
eff
2
R
H
n
2
Z
eff
=
Z
-
σ
\left\{ \begin{aligned} &E_n=-\frac{Z_{\text{eff}}^2 R_H}{n^2} \\ &Z_{\text{eff}}=Z-\sigma \end{aligned} \right.
⎩
⎨
⎧En=−n2Zeff2RHZeff=Z-σ
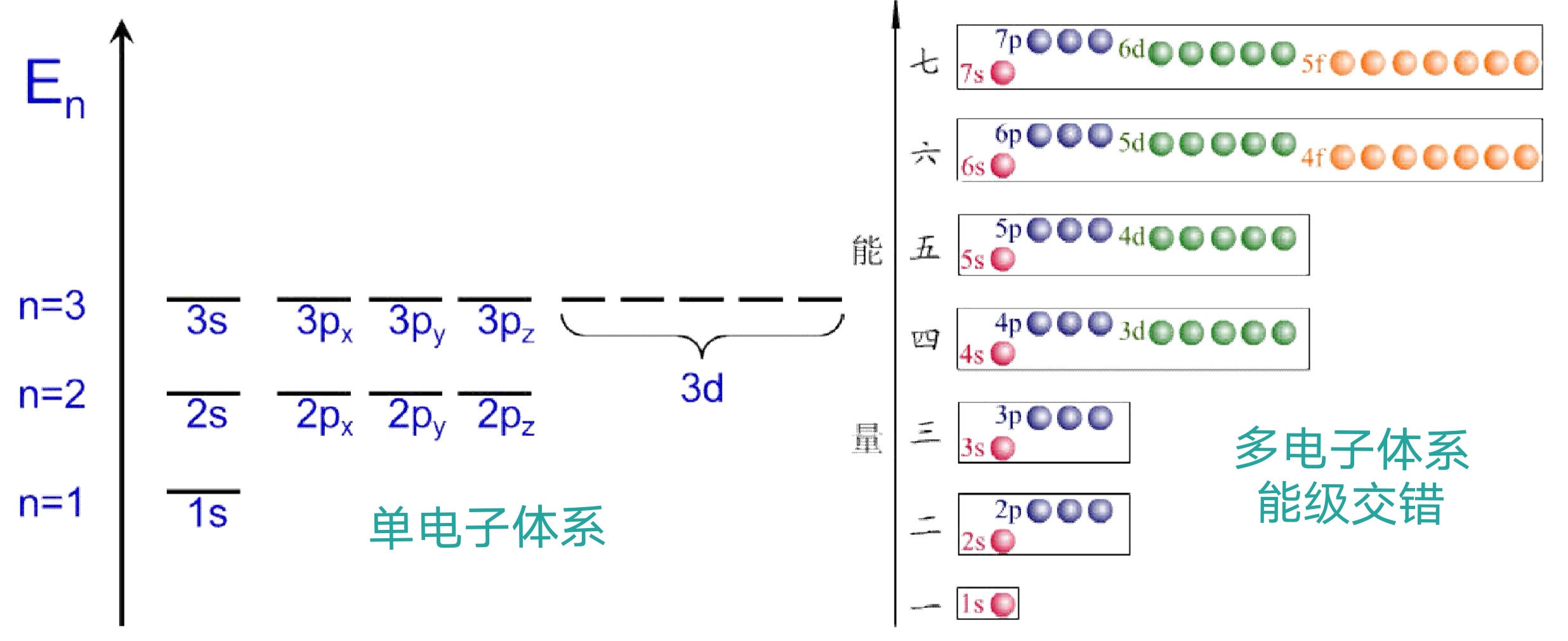
多电子原子轨道能级
能级分裂与能级交错
能级分裂:同一电子层内,轨道能级随角量子数增大而增大
能级交错:不同电子层之间轨道能级的交错分布
屏蔽效应与钻穿效应
-
屏蔽效应:电子之间的相互作用等效为电子在一定程度上抵消了原子核对其它电子的吸引作用
Z eff = Z - σ Z_{\text{eff}}=Z-\sigma Zeff=Z-σ -
钻穿效应:外层电子若出现在核附近的概率较大,则可削弱其它电子对它的屏蔽效应,能量较低,同时对其它电子产生屏蔽。 l l l 越小,钻穿能力越强
用屏蔽效应与钻穿效应解释原子轨道的能级交错
外层角量子数小的轨道4s上的电子钻穿效应强,受到其它电子对屏蔽效应弱,受到原子核吸引力大,能量低,使得 E ( 4s ) < E ( 3d ) E(\text{4s}) < E(\text{3d}) E(4s)<E(3d)
但是该能级交错现象不适用于所有原子,例如 21 Sc {}_{21}\!\text{Sc} 21Sc 之后 E ( 4s ) > E ( 3d ) E(\text{4s})>E(\text{3d}) E(4s)>E(3d)。这些原子中的4s电子受到屏蔽效应占主导地位,这时钻穿效应是次要的。
核外电子排布规则
- 能量最低原理
- 泡利不相容原理
- 洪特规则
元素周期律
原子半径
- 共价半径、金属半径、范德华半径
- 从左到右,主族元素半径减小最快,过渡元素较慢,内过渡元素半径减小最慢(但15个元素总体收缩效应较大,镧系收缩)
电离能
- 同周期从左到右依次增大
- 同族从上到下依次减小
- N, Al \text{N, Al} N, Al 特例:当比半充满或全充满多一个电子时,这种结构不稳定,易失去一个电子变成半充满或全充满状态
- 内过渡元素电离能变化不明显
电子亲和能
- 一般第一亲和能为正,第二亲和能为负
- 从左到右增大,从上到下减小
- 但第2周期从B到F的电子亲和能均低于第3周期同族元素。 这是由于第2周期元素原子半径小,电子云密集导致电子间更强的排斥力,因而形成负离子时放出的能量相对较小。
电负性 χ \chi χ
综合考虑电离能与电子亲和能
表示分子中原子对成键电子的相对吸引力
第2章 分子结构与化学键
离子键
离子键强度
- 表示方法:晶格能
- 100kpa,298K,相互远离的气态正负离子结合成1mol离子晶体释放的能量
- 影响因素
- 离子电荷
- 离子半径(X射线衍射法)
离子极化
离子在电场中原子核与电子云相对位移产生诱导偶极
- 离子的极化能力(产生电场的能力)(阳离子为主)
- 电荷量大、半径小:极化能力强
- 电子构型与极化能力:18(18+2)构型 > 不规则构型 > 8电子构型
- 离子的(电子云)变形性(阴离子为主)
- 半径大:变形性大
- 得电子多:变形性大
- 失电子多:变形性小
- 电子构型与变形性:18构型 > 不规则构型 > 8电子构型
- 离子极化对离子晶体性质的影响
- 由典型离子键向共价键递变
- 物理化学性质递变(极化作用强,颜色深)
共价键
价键理论
- 共价键本质:原子轨道重叠
- 饱和性、方向性(最大重叠原理)
- σ \sigma σ 键, π \pi π 键
- 极性键( Δ χ > 1.7 \Delta \chi>1.7 Δχ>1.7离子键)
- 键能、键长、键角、键矩
- 当分子中有多个相同的键时,键能为这些键离解能的平均值
杂化轨道理论
- 杂化类型
| 杂化类型 | 分子几何构型|
|:------------😐:---------😐
| s p \rm sp sp |直线形 |
| s p 2 \rm sp^2 sp2 |三角形 |
| s p 3 \rm sp^3 sp3 |四面体形 |
| s p 3 d \rm sp^3d sp3d |三角双锥 |
| s p 3 d 2 \rm sp^3d^2 sp3d2 |八面体 | - 不等性杂化
- 斥力大
- 能量低
- 杂化轨道理论解释分子几何构型
- 中心原子价电子构型为 ( 2 s 2 2 p 3 \rm 2s^22p^3 2s22p3)
- 中心原子杂化方式为 ( s p 2 \rm sp^2 sp2)
- (不)等性杂化
- 与…原子形成…个 σ \sigma σ 键,有…对孤电子对
- 所以几何构型为…
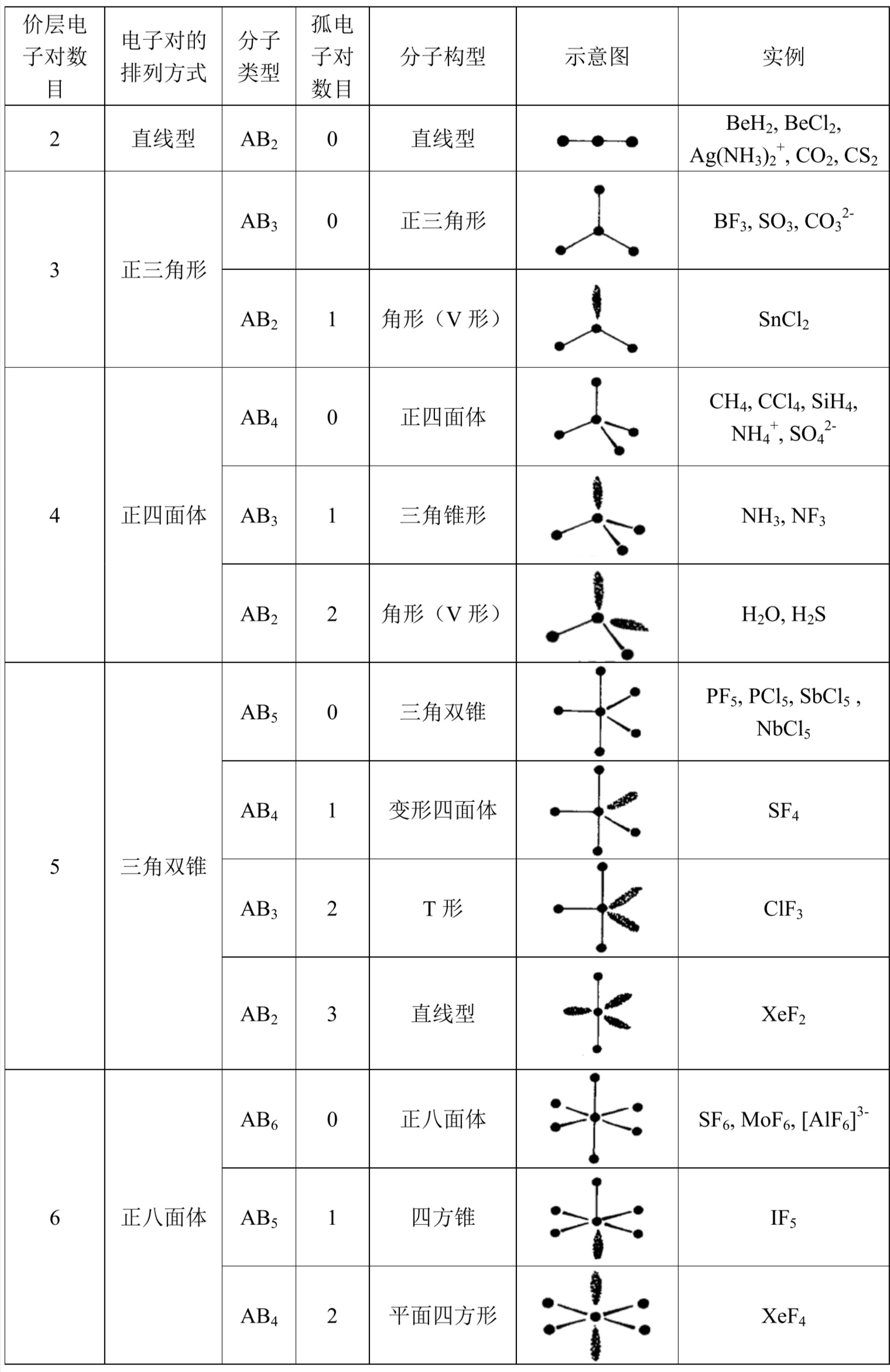
价层电子对互斥理论
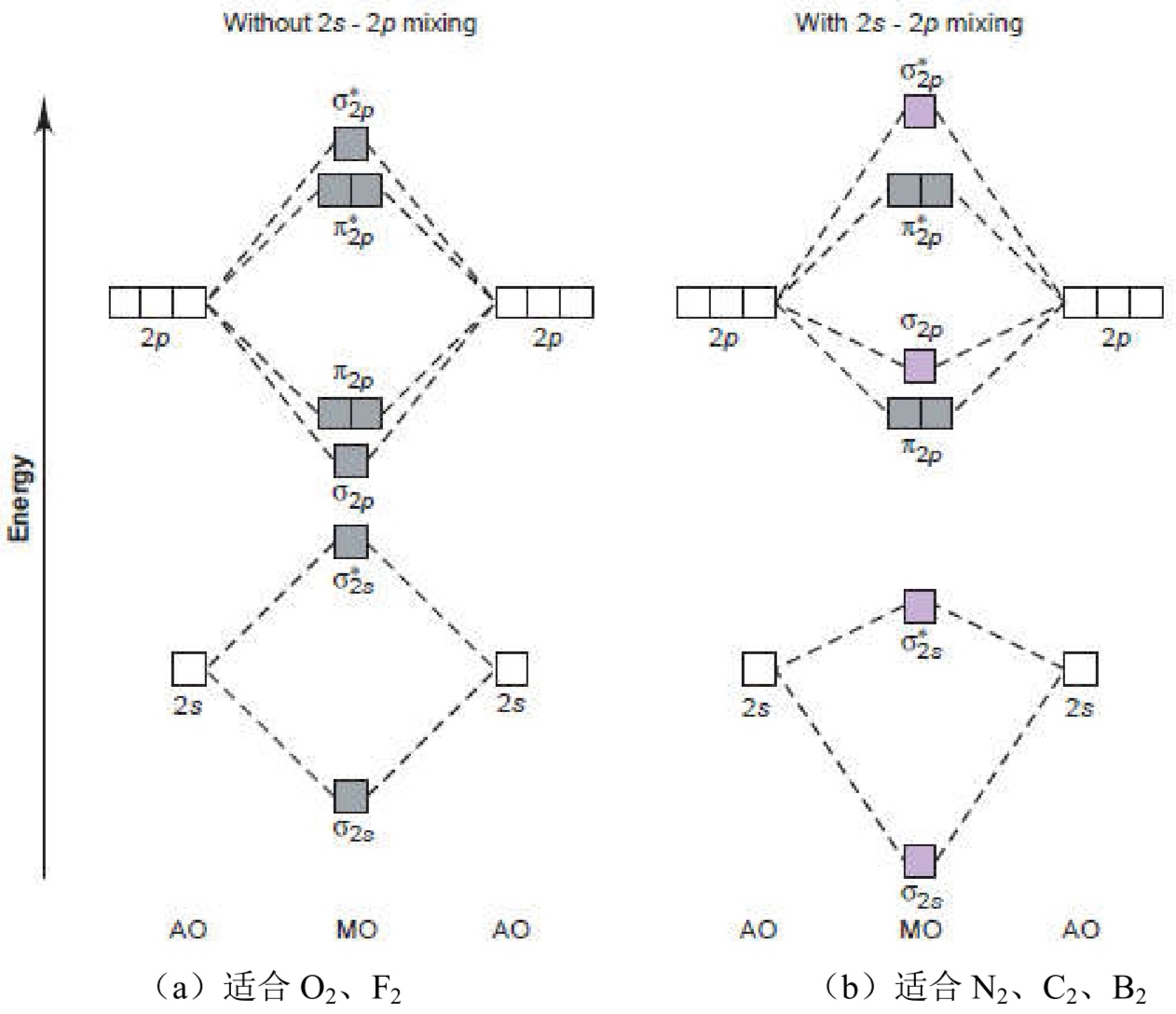
分子轨道理论
- 成键轨道(吸引力):
ψ
1
=
ψ
1
s
+
ψ
1
s
\psi_1=\psi_{1s}+\psi_{1s}
ψ1=ψ1s+ψ1s
反键轨道(排斥力): ψ 1 = ψ 1 s − ψ 1 s \psi_1=\psi_{1s}-\psi_{1s} ψ1=ψ1s−ψ1s - 原子轨道有效组合为分子轨道的三原则
- 对称性匹配原则(首要)
- 能量近似原则
- 最大重叠原则
- 键级=(成键轨道电子数-反键轨道电子数)/2
- 分子轨道能级顺序
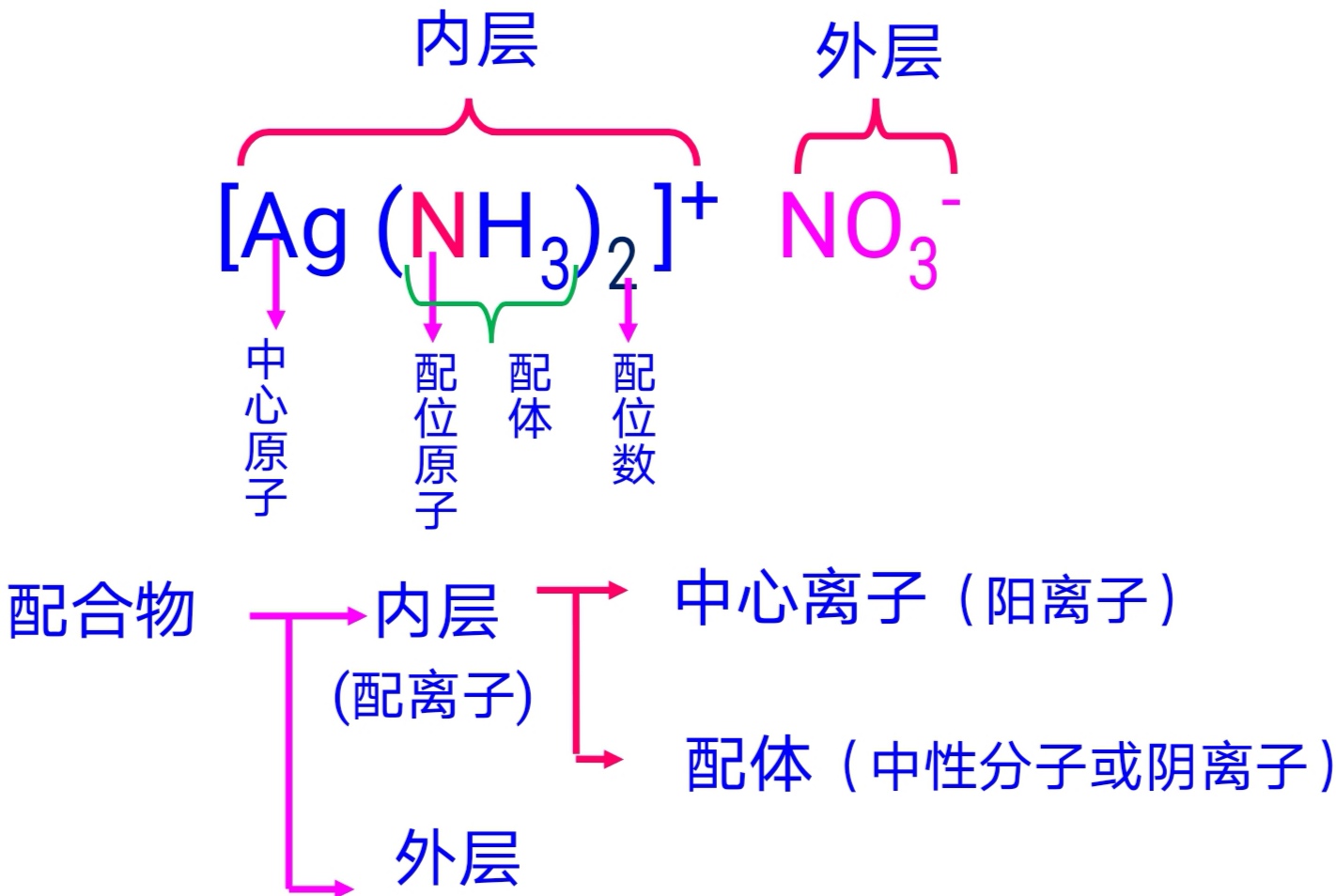
配位键与配位化合物
配合物的组成
| 配体 | 中心原子 |
|---|---|
| N H 3 , N C S − \rm NH_3,NCS^- NH3,NCS− | N \rm N N |
| C N − , C O \rm CN^-,CO CN−,CO(羰) | C \rm C C |
| S C N − , S 2 O 3 2 − \rm SCN^-,S_2O_3^{2-} SCN−,S2O32− | S \rm S S |
配离子的命名
- 阴离子配位体→中性分子配位体→“合”→中心离子(用罗马数字标明氧化数)
- 若有几种阴离子配位体,命名顺序是:简单离子→复杂离子→有机酸根离子。
- 同类配位体按配位原子元素符号英文字母顺序排序。
- 各配位体的个数用数字一、二、三……写在该种配位体名称的前面。不同配位体之间以“•”隔开。

第三章 宏观物质及其聚集态结构
分子间作用力及其聚集状态
分子间力
- 色散力
- 存在于任何分子间
- 一般为分子间力的主要部分
- 分子量大,色散力大
- 分子体积大,色散力大
- 分子结构松散,变形性大,色散力大
- 诱导力
- 存在于极性-非极性、极性-极性分子间
- 大小与极性分子的极性与非极性分子的变形性有关
- 取向力
- 极性-极性
- 分子极性大,取向力大
- 有氢键时显著增大
- 氢键
- 分子内氢键使熔沸点下降,蒸汽压下降
- 分子间氢键:
- 熔沸点升高,蒸汽压增大
- 粘度增大
- 酸性减弱(氢氟酸)
液体
蒸汽压
- 一定温度下,真空容器中,液体蒸发的速率与蒸汽凝结的速率相等时,气液两相达到平衡,此时蒸汽的压强被称为溶剂的(饱和)蒸气压
- 温度升高,蒸气压大
- 分子间作用力大,蒸气压小
相图
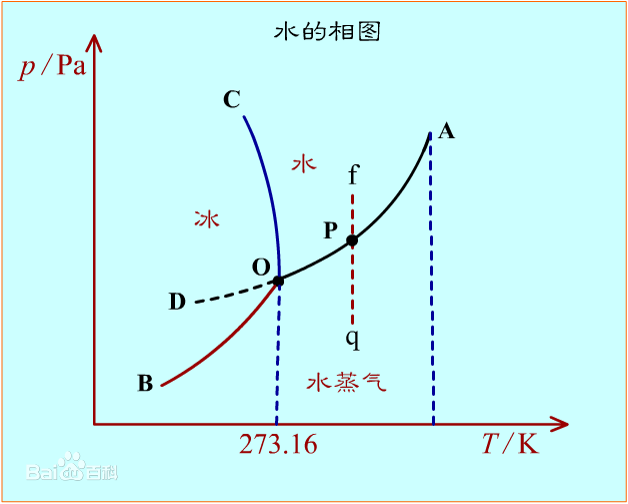
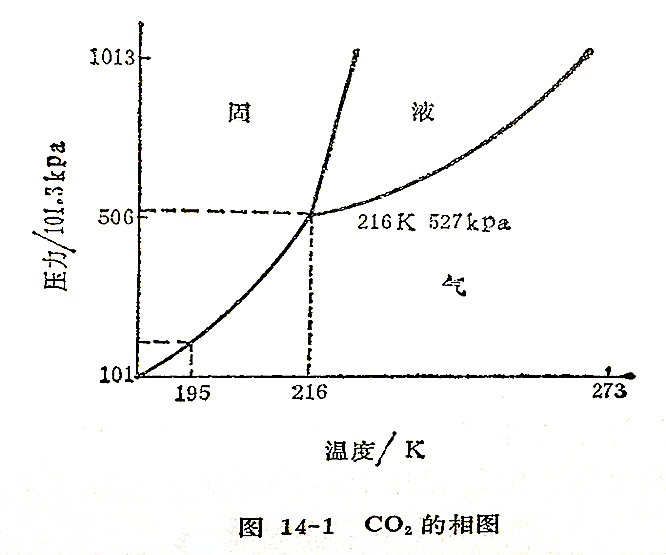
溶液
分散系及其分类
-
分散质(分散相)
-
分散剂(分散介质)
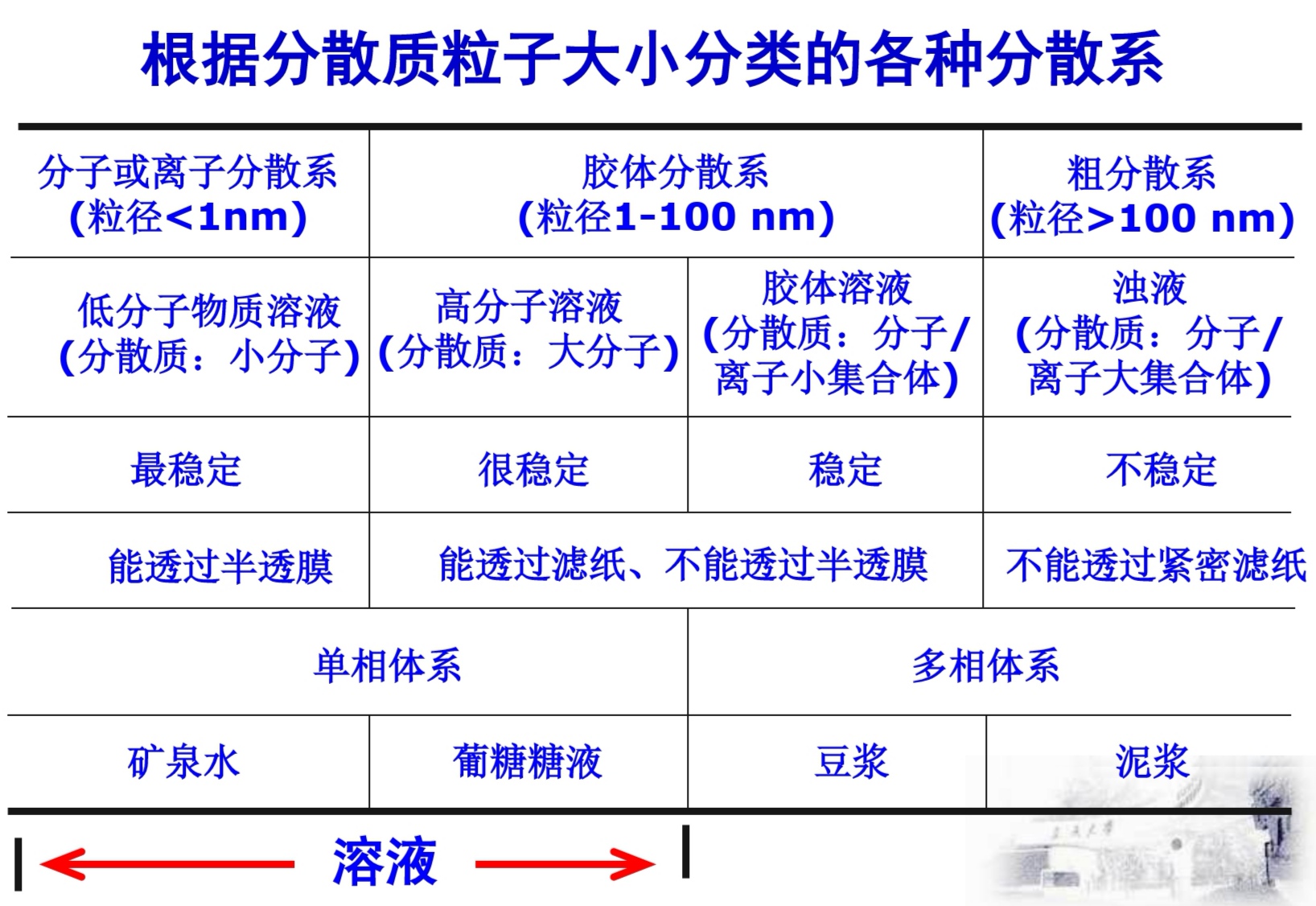
溶液浓度的表征
- 分数
- 质量分数 ω \omega ω
- 体积分数 φ \varphi φ
- 物质的量分数 x x x
- 体积摩尔浓度
c
c
c,
mol/L - 质量摩尔浓度
m
m
m,
mol/kg1kg溶剂中溶质的物质的量
稀溶液的依数性
难挥发非电解质稀溶液的依数性
-
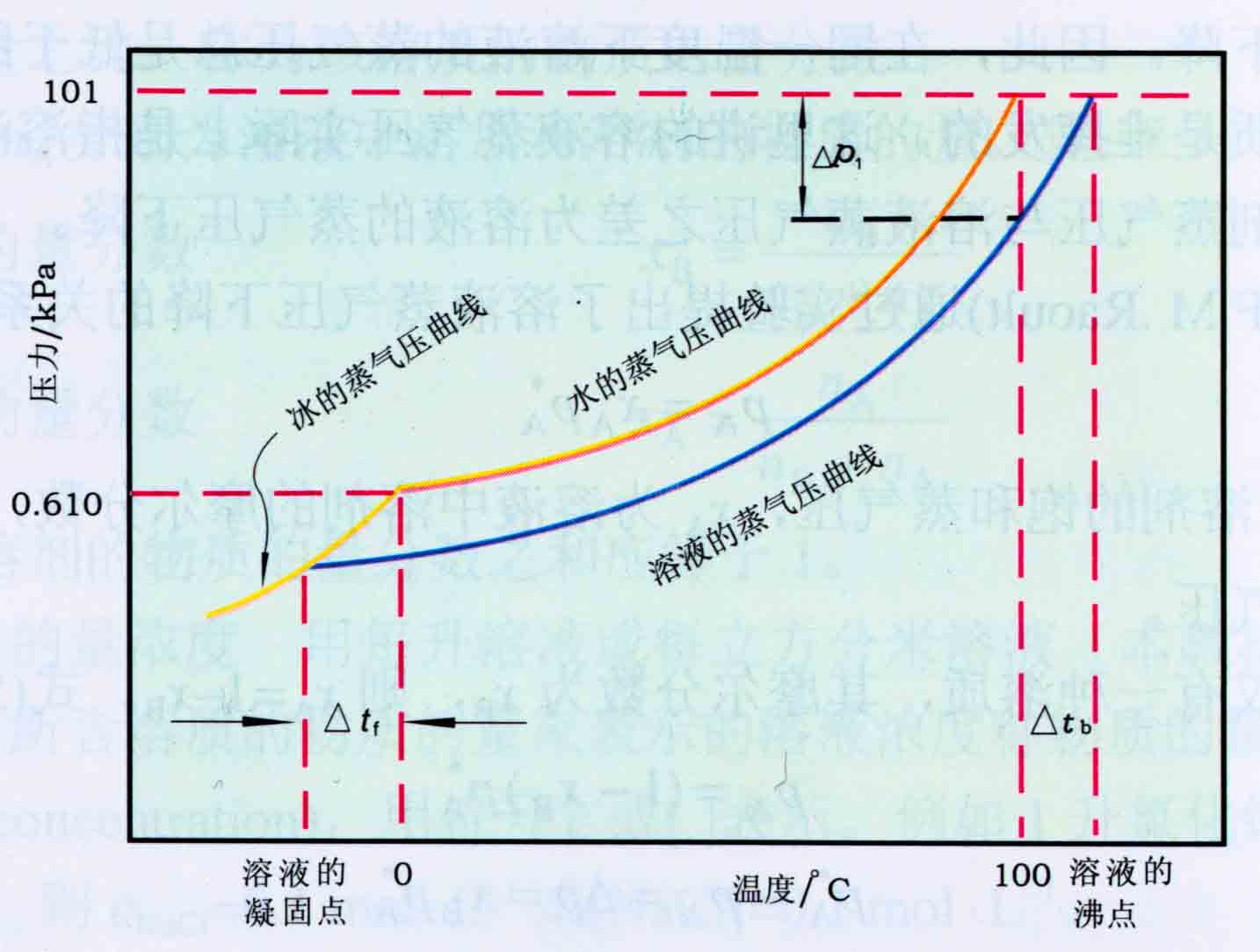
蒸气压下降
p A − p = Δ p = x B p A = n B n A + n B p A ≈ n B n A p A = n B w A / M A p A = K c m B p_A-p=\Delta p=x_Bp_A=\frac{n_B}{n_A+n_B}p_A\approx \frac{n_B}{n_A}p_A=\frac{n_B}{w_A/M_A}p_A=K_cm_B pA−p=Δp=xBpA=nA+nBnBpA≈nAnBpA=wA/MAnBpA=KcmB -
沸点升高,凝固点下降
- 沸点:随温度上升,液体蒸气压增大到与外界压强相同
- 凝固点:随温度下降,液体蒸气压下降到与此温度下固体蒸气压相等
- 沸点、凝固点变化与溶质质量摩尔浓度成正比
{ Δ t b = K b m B Δ t f = K f m B \left\{ \begin{aligned} &\Delta t_b=K_bm_B\\ &\Delta t_f=K_fm_B \end{aligned} \right. {Δtb=KbmBΔtf=KfmB
- 有渗透压
Π V = n R T \Pi V=nRT ΠV=nRT
晶体
- 离子晶体
- 分子晶体
- 原子晶体
- 金属晶体
- 混合型晶体
液晶
由固态向液态转化过程中存在的取向有序流体
第四章 化学热力学
热力学能、反应热、焓
热力学能
{ Δ U = Q + W W = − p Δ V \left\{\begin{aligned} & \Delta U = Q + W \\ & W = -p \Delta V \end{aligned}\right . {ΔU=Q+WW=−pΔV
反应热 Q Q Q
体系不做非体积功、反应前后温度相等时,所吸收的热量
- 等压热效应 Q p = Δ H Q_p = \Delta H Qp=ΔH
- 等容热效应 Q V = Δ U Q_V = \Delta U QV=ΔU
焓
体系不做非体积功,恒压过程
{
p
Δ
V
=
Δ
n
R
T
H
=
U
+
p
V
Q
p
=
Δ
H
Q
V
=
Δ
U
Q
p
−
Q
V
=
Δ
H
−
Δ
U
=
p
Δ
V
=
Δ
n
R
T
\left\{ \begin{aligned} &p\Delta V=\Delta nRT\\ &H=U+pV\\ &Q_p=\Delta H\\ &Q_V=\Delta U\\ &Q_p - Q_V=\Delta H-\Delta U=p\Delta V=\Delta nRT\\ \end{aligned}\right .
⎩
⎨
⎧pΔV=ΔnRTH=U+pVQp=ΔHQV=ΔUQp−QV=ΔH−ΔU=pΔV=ΔnRT
标准状态下的焓与熵
标准状态
p
Θ
=
100
k
P
a
p^\Theta=100\ {\rm kPa}
pΘ=100 kPa
溶液中
c
Θ
=
1
m
o
l
/
L
c^\Theta=1\ {\rm mol/L}
cΘ=1 mol/L 或
m
Θ
=
1
m
o
l
/
k
g
m^\Theta=1\ {\rm mol/kg}
mΘ=1 mol/kg
无温度要求
标准焓变
标准状态,温度为 T T T 时
- 标准摩尔焓变 Δ r H m Θ \Delta_r H_m^\Theta ΔrHmΘ
- 标准摩尔生成焓
Δ
f
H
m
Θ
\Delta_f H_m^\Theta
ΔfHmΘ
- 由参考态单质生成1摩尔化合物
- 标准摩尔燃烧焓 Δ c H m Θ \Delta_c H_m^\Theta ΔcHmΘ
标准熵变
标准状态,温度为
T
T
T时
标准摩尔熵变
Δ
S
m
Θ
\Delta S_m^\Theta
ΔSmΘ
Q r = T Δ S Q_r=T\Delta S Qr=TΔS:代表过程中因熵变而需要吸收或放出的最小热量
Gibbs函数 G = H − T S G=H-TS G=H−TS
等温状态下,Gibbs函数变 Δ G = Δ H − T Δ S \Delta G=\Delta H-T\Delta S ΔG=ΔH−TΔS
Gibbs函数的物理意义
Δ U = Q − p Δ V + W Q m i n = Q r = T Δ S } ⇒ Δ U + p Δ V = T Δ S + W m a x Δ H = Δ U + p Δ V Δ H = Δ G + T Δ S } ⇒ Δ G = W m a x \left . \begin{aligned} \left . \begin{aligned} \Delta U=Q-p\Delta V+W&\\ Q_{min}=Q_r=T\Delta S& \end{aligned} \right\} \Rightarrow \Delta U+p\Delta V=T\Delta S+W_{max}&\\ \Delta H=\Delta U+p\Delta V&\\ \Delta H=\Delta G+T\Delta S& \end{aligned} \right\} \Rightarrow \Delta G=W_{max} ΔU=Q−pΔV+WQmin=Qr=TΔS}⇒ΔU+pΔV=TΔS+WmaxΔH=ΔU+pΔVΔH=ΔG+TΔS⎭ ⎬ ⎫⇒ΔG=Wmax
即: Δ G \Delta G ΔG代表理论上可以利用的最大非体积功。若不能有效利用,则也转化为热量。
标准Gibbs函数变
Δ r G m , T Θ = Δ r H m , T Θ − T Δ r S m , T Θ ≈ Δ r H m , 298 Θ − T Δ r S m , 298 Θ \Delta_r G_{m,T}^\Theta = \Delta_r H_{m,T}^\Theta - T\Delta_r S_{m,T}^\Theta \approx \Delta_r H_{m,298}^\Theta - T\Delta_r S_{m,298}^\Theta ΔrGm,TΘ=ΔrHm,TΘ−TΔrSm,TΘ≈ΔrHm,298Θ−TΔrSm,298Θ
标准摩尔生成Gibbs函数变 Δ f G m Θ \Delta_f G_m^\Theta ΔfGmΘ
第五章 化学平衡
标准平衡常数与反应商
标准平衡常数
K
Θ
K^\Theta
KΘ 只与温度有关
当达到平衡时,反应商
J
=
K
Θ
J=K^\Theta
J=KΘ
多重平衡法则
对于化学反应
A
,
B
,
C
A,B,C
A,B,C
C
=
m
A
+
n
B
⇒
K
C
Θ
=
(
K
A
Θ
)
m
⋅
(
K
B
Θ
)
n
C=mA+nB \Rightarrow K_C^\Theta=(K_A^\Theta)^m \cdot (K_B^\Theta)^n
C=mA+nB⇒KCΘ=(KAΘ)m⋅(KBΘ)n
非标准状态下的Gibbs函数变
{ Δ r G m , T = Δ r G m , T Θ + R T ln J Δ r G m , T Θ + R T ln K Θ = 0 Δ r G m , T = R T ln J / K Θ \left\{ \begin{aligned} &\Delta_rG_{m,T}=\Delta_rG_{m,T}^\Theta+RT\ln J \\ &\Delta_rG_{m,T}^\Theta+RT\ln {K^\Theta}=0 \\ &\Delta_rG_{m,T} = RT\ln { J /{K^\Theta}} \end{aligned} \right. ⎩ ⎨ ⎧ΔrGm,T=ΔrGm,TΘ+RTlnJΔrGm,TΘ+RTlnKΘ=0ΔrGm,T=RTlnJ/KΘ
其中
Δ
r
G
m
,
T
\Delta_rG_{m,T}
ΔrGm,T 的值随反应程度变化
Δ
r
G
m
,
T
Θ
\Delta_rG_{m,T}^\Theta
ΔrGm,TΘ 是反应各反应物刚好都处于标准状态下时
Δ
r
G
m
,
T
\Delta_rG_{m,T}
ΔrGm,T 的特殊取值
由于不同温度下的
Δ
r
G
m
,
T
Θ
\Delta_rG_{m,T}^\Theta
ΔrGm,TΘ 与
K
Θ
K^\Theta
KΘ 一一对应,
Δ
r
G
m
,
T
Θ
\Delta_rG_{m,T}^\Theta
ΔrGm,TΘ 可以代表化学反应进行的限度
平衡移动
温度对化学平衡的影响
T → influence Δ r G m , T Θ ⇔ K Θ T \xrightarrow {\text{influence}}\Delta_rG_{m,T}^\Theta \Leftrightarrow K^\Theta TinfluenceΔrGm,TΘ⇔KΘ
范特霍夫方程式
−
R
T
1
ln
K
1
Θ
=
Δ
r
G
m
,
T
1
Θ
=
Δ
r
H
m
,
T
1
Θ
−
T
1
Δ
r
S
m
,
T
1
Θ
−
R
T
2
ln
K
2
Θ
=
Δ
r
G
m
,
T
2
Θ
=
Δ
r
H
m
,
T
2
Θ
−
T
2
Δ
r
S
m
,
T
2
Θ
}
⇒
ln
K
1
Θ
K
2
Θ
=
Δ
r
H
m
Θ
R
(
1
T
2
−
1
T
1
)
\left. \begin{aligned} &-RT_1\ln K_1^\Theta=\Delta_rG_{m,T_1}^\Theta=\Delta_r H^\Theta_{m,T_1}-T_1 \Delta_r S_{m,T_1}^\Theta \\ &-RT_2\ln K_2^\Theta=\Delta_rG_{m,T_2}^\Theta=\Delta_r H^\Theta_{m,T_2}-T_2\Delta_r S_{m,T_2}^\Theta \\ \end{aligned} \right\} \\ \Rightarrow \ln{\frac {K_1^\Theta}{K_2^\Theta}}=\frac{\Delta_r H_m^\Theta}{R}\left( \frac 1 {T_2} - \frac 1 {T_1}\right)
−RT1lnK1Θ=ΔrGm,T1Θ=ΔrHm,T1Θ−T1ΔrSm,T1Θ−RT2lnK2Θ=ΔrGm,T2Θ=ΔrHm,T2Θ−T2ΔrSm,T2Θ}⇒lnK2ΘK1Θ=RΔrHmΘ(T21−T11)
上式中认为焓变、熵变受温度影响较小
压强、浓度对化学平衡的影响
通过反应商 J J J 确定
当分压压强等比变化
n
n
n 时(假设原先处于平衡状态)
a
A
(
g
)
+
b
B
(
g
)
⇌
c
C
(
g
)
J
=
n
c
n
b
n
a
J
0
=
n
c
−
b
−
a
K
Θ
\begin{aligned} aA(\text{g})+bB(\text{g}) \rightleftharpoons cC(\text{g})\\ J=\frac{n^c}{n^bn^a}J_0=n^{c-b-a}K^\Theta \end{aligned}
aA(g)+bB(g)⇌cC(g)J=nbnancJ0=nc−b−aKΘ
即:压缩时平衡向分子数减小的方向移动
浓度同理
第6章 水溶液中的离子平衡
强电解质溶液理论
解离度 α \alpha α
α = 已解离分子数 原有分子总数 × 100 % \alpha = \frac{已解离分子数}{原有分子总数} \times 100\% α=原有分子总数已解离分子数×100%
α \alpha α 的值可以通过测量依数性 Δ p , Δ t f , Δ t b , Π \Delta p,\; \Delta t_f,\; \Delta t_b,\; \Pi Δp,Δtf,Δtb,Π 得到
对于强电解质溶液,正负离子的相互作用,在每个离子附近球形区域内吸引一些带有相反电荷的离子,形成离子氛。
离子氛制约了溶液中离子的自由移动,使得实验中测得的解离度小于理论值,称为表观解离度
强电解质的极稀溶液中,表观解离度接近100%
离子活度 a a a 与活度因子 γ \gamma γ
离子活度
a
a
a 为溶液中离子的有效相对浓度
无单位的纯数
活度因子
γ
\gamma
γ:
γ
=
a
c
\gamma = \frac a c
γ=ca
离子活度与理论相对浓度的比值 γ < 1 \gamma<1 γ<1
影响离子活度与活度因子的主要因素有溶液浓度与离子电荷量
溶液浓度越大,离子间牵制作用越强,离子氛出现机会越多,活度因子越小
弱电解质稀溶液、难溶电解质溶液、中性分子溶液 γ ≈ 1 \gamma \approx 1 γ≈1
无法单独测量溶液中正或负离子的活度、活度因子,只能测量其平均值。
对于
1
:
1
1:1
1:1型离子化合物:
{
γ
±
=
γ
+
γ
−
a
±
=
a
+
a
−
\begin{cases} \gamma_{\pm} = \sqrt{\gamma_+ \gamma_-} \\ a_{\pm}=\sqrt{a_+a_-} \end{cases}
{γ±=γ+γ−a±=a+a−
离子强度 I I I
描述离子间作用力的强度
I
=
1
2
∑
i
m
i
z
i
2
I = \frac 1 2 \sum_i{m_iz_i^2}
I=21i∑mizi2
其中 m i m_i mi 为离子的质量摩尔浓度, z i z_i zi为离子电荷量
离子强度 I I I 越大,离子间作用力越强,离子活度 a a a 与活度因子 γ \gamma γ 越小
溶液浓度
c
<
0.01
m
o
l
/
L
c < 0.01 \rm mol/L
c<0.01mol/L 时,
lg
γ
±
=
−
0.509
∣
z
−
z
+
∣
I
\lg{\gamma_{\pm} = -0.509|z_- z_+|\sqrt{I}}
lgγ±=−0.509∣z−z+∣I
溶液浓度
c
<
0.01
m
o
l
/
L
c < 0.01 \rm mol/L
c<0.01mol/L 时,
lg
γ
±
=
−
0.509
∣
z
−
z
+
∣
I
1
+
I
\lg{\gamma_{\pm}} = \frac{-0.509|z_- z_+|\sqrt{I}}{1+ \sqrt{I}}
lgγ±=1+I−0.509∣z−z+∣I
盐效应
向弱电解质或难溶电解质溶液中加入无相同离子的强电解质,由于离子总浓度增大,离子间相互牵制增强,原溶质的阴阳离子活度降低,平衡正移,使得弱电解质解离度增大、难溶电解质溶解度增强
酸碱理论
酸碱电离理论
酸:水中电离出的阳离子均为
H
+
\text{H}^+
H+ 的物质
碱:水中电离出的阴离子全部为
OH
−
\text{OH}^-
OH− 的物质
酸碱反应的实质:
H
+
+
O
H
−
=
H
2
O
\rm H^+ + OH^- \xlongequal{\quad} H_2O
H++OH−H2O
优点1:可以定量描述酸碱强弱
缺陷1:无法说明非水溶液中的酸碱反应
缺陷2:无法解释弱酸弱碱盐的酸碱性
酸碱质子理论
酸:给出质子的物质
碱:接受质子的物质
酸碱反应的实质:强碱夺取强酸的质子,生成弱酸、弱碱
酸给出质子变为碱,碱接受质子变为酸,成为一对共轭酸碱
如:
H
3
O
+
⇌
H
+
+
H
2
O
\rm H_3O^+ \rightleftharpoons H^+ + H_2O
H3O+⇌H++H2O
酸碱强弱:同一溶剂中给出或接受质子的能力
既与酸碱物质的本性有关,又与溶剂的性质有关
酸越强,其共轭碱越弱
某溶剂中的最强酸:溶剂合质子(
H
3
O
+
\rm H_3O^+
H3O+)
某溶剂中的最强碱:溶剂阴离子(
O
H
−
)
\rm OH^-)
OH−)
优点1:扩大了酸碱的范围
优点2:扩大了酸碱反应的范围
优点3:消除了盐的概念
缺点1:对于不含质子的物质,无法解释
酸碱电子理论
酸:接受电子对的物质
碱:给出电子对的物质
酸碱反应的实质:电子对接收体与给予体形成配位共价键
酸碱反应的分类:
| 酸碱反应 | 反应式 |
|---|---|
| 酸碱加和反应 | A + : B → A − B \rm A\;+:\!\!B \to A\!\!-\!\!B A+:B→A−B |
| 酸取代反应 | A − B + A ′ → A ′ − B + A \rm A\!\!-\!\!B+A'\to A'\!\!-\!\!B+A A−B+A′→A′−B+A |
| 碱取代反应 | A − B + B ′ → A − B ′ + B \rm A\!\!-\!\!B+B'\to A\!\!-\!\!B'+B A−B+B′→A−B′+B |
| 双取代反应 | A − B + A ′ − B ′ → A ′ − B + A ′ − B \rm A\!\!-\!\!B+A'\!\!-\!\!B'\to A'\!\!-\!\!B+A'\!\!-\!\!B A−B+A′−B′→A′−B+A′−B |
水的解离平衡
质子溶剂:既可以作为酸,又可以作为碱的溶剂
质子自递反应:质子溶剂分子之间发生质子转移
K
w
Θ
=
[
H
+
]
⋅
[
OH
−
]
K_{\text w}^\Theta=[\text{H}^+]\cdot[\text{OH}^-]
KwΘ=[H+]⋅[OH−]
298
K
298 \; \text K
298K 下
{
K
w
Θ
=
1
0
−
14
pH
+
pOH
=
p
K
w
Θ
=
14
\begin{cases} K_{\text w}^\Theta = 10^{-14}\\ \text p\text{H} + \text p\text{OH} = \text pK_{\text w}^\Theta = 14 \end{cases}
{KwΘ=10−14pH+pOH=pKwΘ=14
单相离子平衡
电解质解离前后相态相同的解离平衡
一元弱酸弱碱的解离平衡
{ c ( H + ) ≈ K a Θ c a α = c ( H + ) c a = K a Θ / c a \begin{cases} c\left( \text H ^+\right) \approx \sqrt{K_a^\Theta c_a} \\ \alpha = \dfrac{c\left( \text H ^+\right)}{c_a} = \sqrt{K_a^\Theta /c_a} \end{cases} ⎩ ⎨ ⎧c(H+)≈KaΘcaα=cac(H+)=KaΘ/ca
对于一对共轭酸碱
H
A
=
H
+
+
A
−
K
a
Θ
A
−
+
H
2
O
=
H
A
+
O
H
−
K
b
Θ
⇒
K
a
Θ
⋅
K
b
Θ
=
K
w
Θ
\begin{aligned} {\rm HA }&\xlongequal{\quad} {\rm H^++A^-} &K_a^\Theta\\ {\rm A^- + H_2O} &\xlongequal{\quad}{\rm HA+OH^-} &K_b^\Theta \end{aligned} \\ \Rightarrow \boxed{K_a^\Theta\cdot K_b^\Theta = K_{\text w}^\Theta}
HAA−+H2OH++A−HA+OH−KaΘKbΘ⇒KaΘ⋅KbΘ=KwΘ
多元弱酸解离平衡
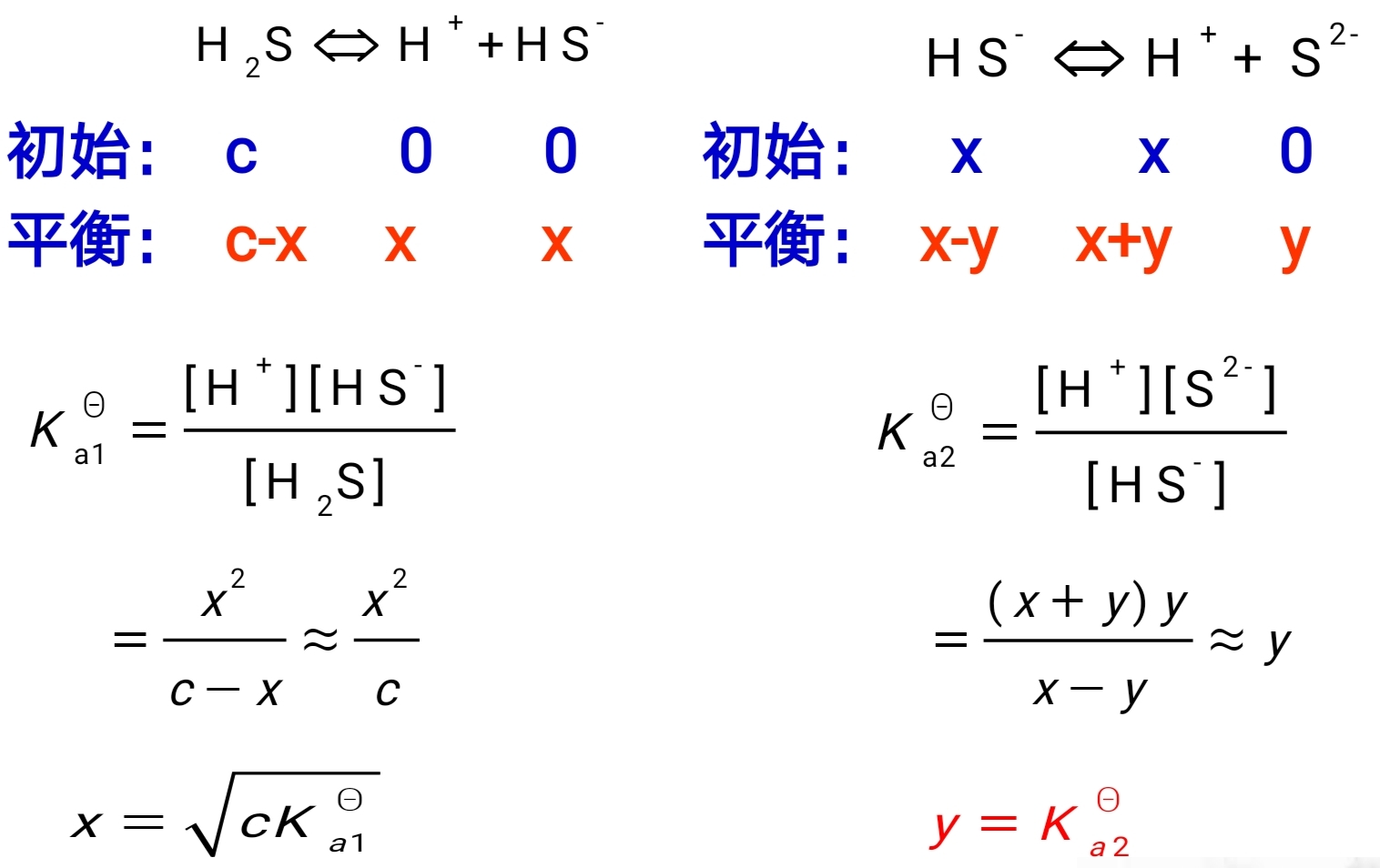 $$ \begin{cases} c\left( \text H ^+\right) \approx \sqrt{K_{a1}^\Theta c_a} \\ c\left( \text A ^{2-}\right) \approx K_{a2}^\Theta \end{cases} $$
$$ \begin{cases} c\left( \text H ^+\right) \approx \sqrt{K_{a1}^\Theta c_a} \\ c\left( \text A ^{2-}\right) \approx K_{a2}^\Theta \end{cases} $$
缓冲溶液
利用同离子效应
缓冲溶液的 pH \text p\text H pH
∵ K a Θ = c ( H + ) c ( A − ) c ( HA ) ≈ c ( H + ) c ( 盐 ) c ( 酸 ) ∴ pH = p K a Θ + lg c ( 盐 ) c ( 酸 ) \begin{aligned} &\because K_a^\Theta=\dfrac{c\left( \text H ^+\right)c\left( \text A ^-\right)}{c\left( \text {HA} \right)} \approx \dfrac{c\left( \text H ^+\right)c\left( \text 盐\right)}{c\left( \text {酸} \right)} \\ &\therefore \boxed{\text p\text H = \text pK_a^\Theta {\color{red}+}\lg{\dfrac{c\left( \text 盐\right)}{c\left( \text {酸} \right)}}} \end{aligned} ∵KaΘ=c(HA)c(H+)c(A−)≈c(酸)c(H+)c(盐)∴pH=pKaΘ+lgc(酸)c(盐)
缓冲能力来源于 c ( 盐 ) c\left( \text 盐\right) c(盐), c ( 酸 ) c\left( \text 酸\right) c(酸)较大使得 lg c ( 盐 ) c ( 酸 ) \lg{\tfrac{c\left( \text 盐\right)}{c\left( \text {酸} \right)}} lgc(酸)c(盐)变化缓慢
缓冲容量
对于 1 L 1 \; \text L 1L 缓冲溶液,升高或降低单位 pH \text p\text H pH 所需要的酸或碱的量
缓冲范围
pH
=
p
K
a
Θ
±
1
\text p\text H = \text pK_a^\Theta \pm 1
pH=pKaΘ±1
c ( 盐 ) c ( 酸 ) > 10 \tfrac{c\left( \text 盐\right)}{c\left( \text {酸} \right)}>10 c(酸)c(盐)>10 或 c ( 盐 ) c ( 酸 ) < 0.1 \tfrac{c\left( \text 盐\right)}{c\left( \text {酸} \right)}<0.1 c(酸)c(盐)<0.1 时,无缓冲能力
最大缓冲能力点
{
c
(
盐
)
=
c
(
酸
)
pH
=
p
K
a
Θ
\begin{cases} c\left( \text 盐\right)=c\left( \text 酸\right) \\ \text p\text H=\text pK_a^\Theta \end{cases}
{c(盐)=c(酸)pH=pKaΘ
增大缓冲容量:增大 c ( 盐 ) c\left( \text 盐\right) c(盐), c ( 酸 ) c\left( \text 酸\right) c(酸)
选定缓冲体系:选择最接近的 p K a Θ pK_a^\Theta pKaΘ
酸碱滴定法
指示剂的变色范围
pH = p K HIn Θ ± 1 \text{pH} = \text pK_{\text{HIn}}^\Theta \pm 1 pH=pKHInΘ±1
滴定突跃
化学计量点前后0.1%溶液pH突跃的现象
相应的pH变化范围为滴定突跃范围
突跃范围影响因素:浓度
浓度增大
⇒
\;\Rightarrow\;
⇒ 滴定突跃范围增大
弱酸弱碱的滴定
弱酸弱碱可以被强酸强碱直接滴定的条件
K
a
Θ
c
a
>
1
0
−
8
K
b
Θ
c
b
>
1
0
−
8
K_a^\Theta c_a > 10^{-8} \\ K_b^\Theta c_b > 10^{-8}
KaΘca>10−8KbΘcb>10−8
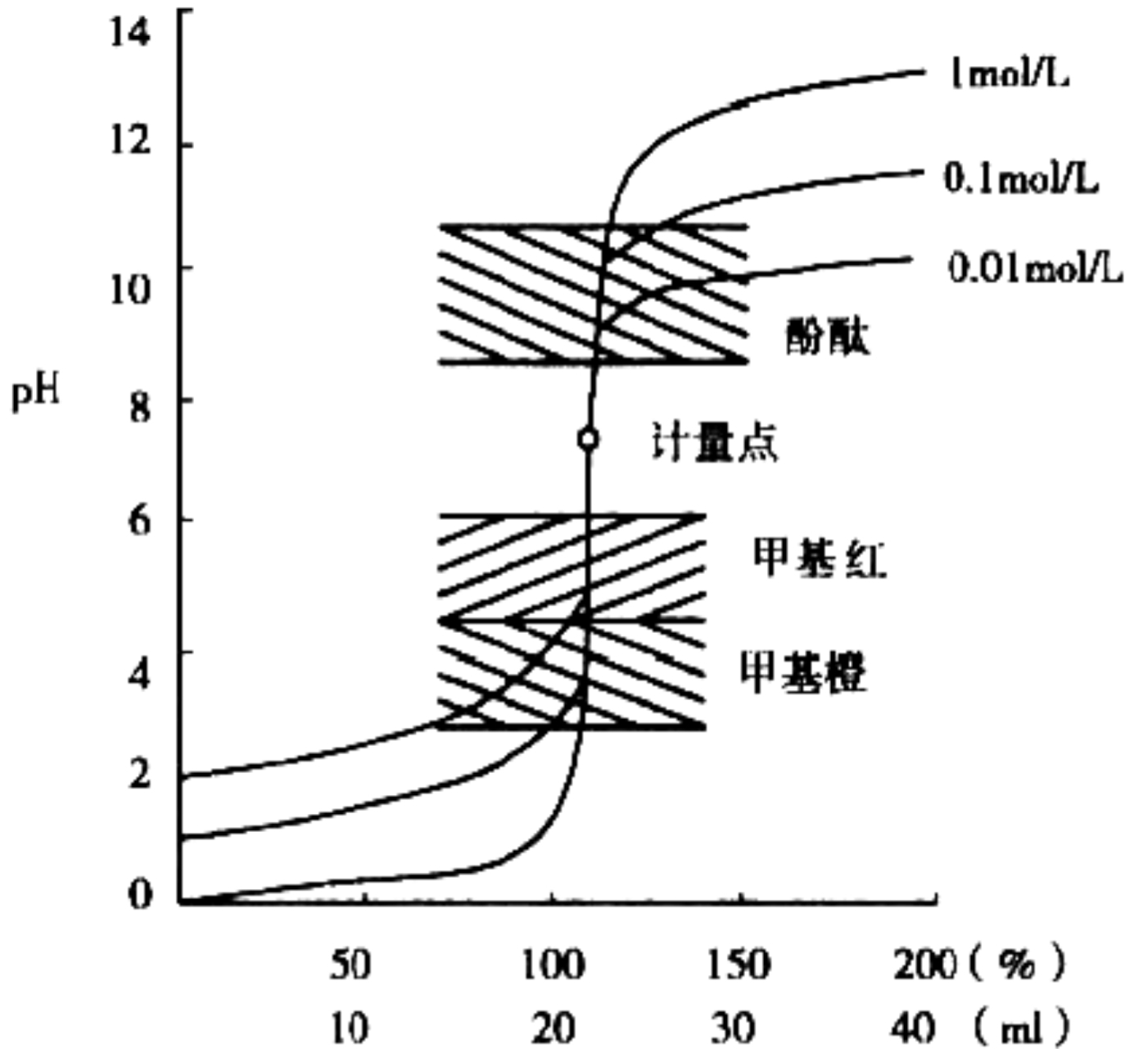
滴定曲线
强碱滴定强酸
强碱滴定弱酸(含有缓冲区)
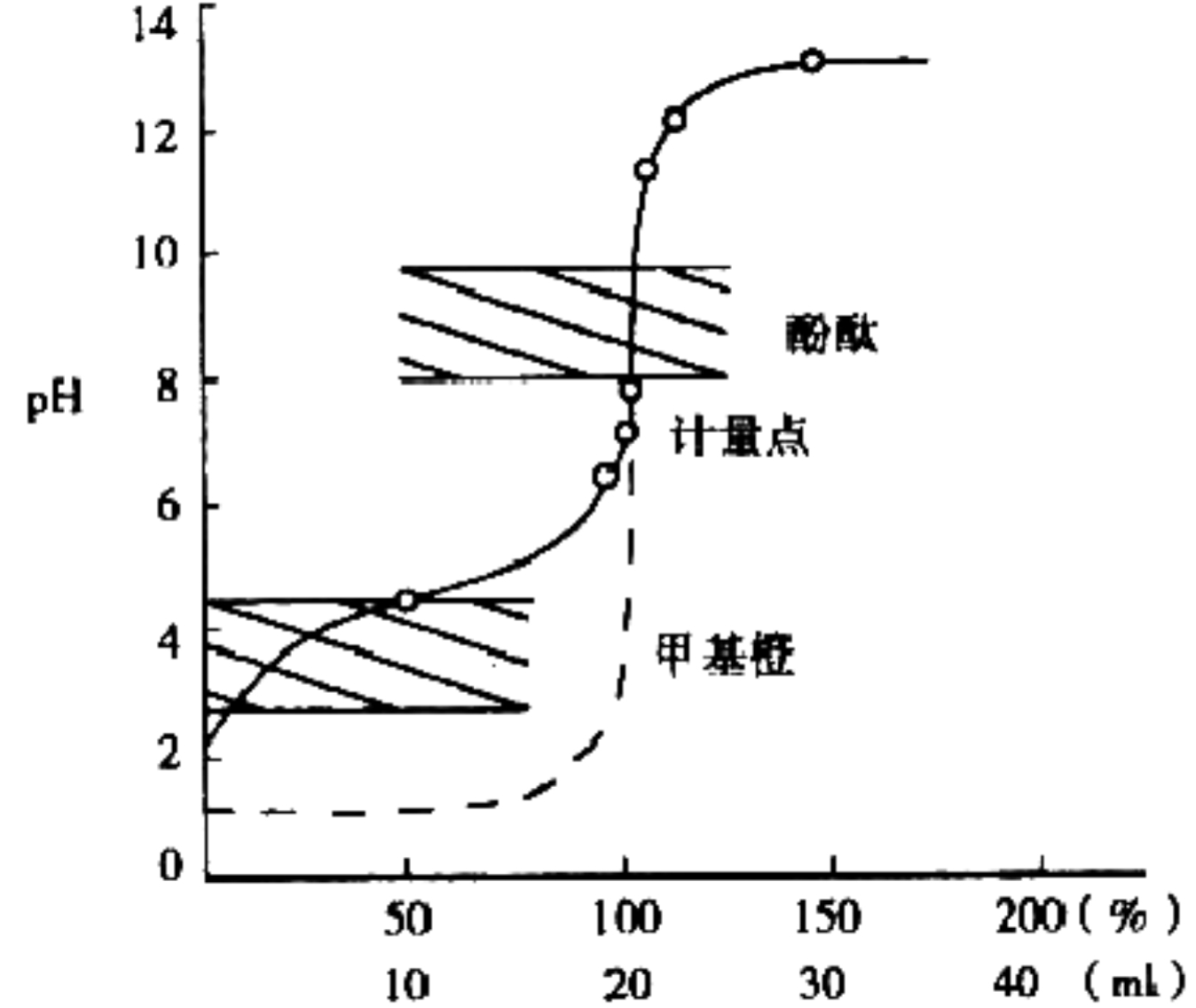
多相离子平衡
溶度积 K sp Θ K_{\text {sp}}^\Theta KspΘ
溶度积规则
比较
J
J
J 与
K
sp
Θ
K_{\text {sp}}^\Theta
KspΘ 的大小
配位平衡
稳定平衡常数:配合物生成反应
A
g
+
+
N
H
3
⇌
[
A
g
(
N
H
3
)
]
+
K
稳
1
Θ
[
A
g
(
N
H
3
)
]
+
+
N
H
3
⇌
[
A
g
(
N
H
3
)
2
]
+
K
稳
2
Θ
\begin{aligned} {\rm Ag^+ + NH_3 }&\rightleftharpoons {\rm [Ag(NH_3)]^+} &K_{\text 稳1}^\Theta \\ {\rm [Ag(NH_3)]^+ + NH_3} &\rightleftharpoons {\rm [Ag(NH_3)_2]^+} &K_{\text 稳2}^\Theta \end{aligned}
Ag++NH3[Ag(NH3)]++NH3⇌[Ag(NH3)]+⇌[Ag(NH3)2]+K稳1ΘK稳2Θ
不稳定平衡常数:配合物分解反应
[
A
g
(
N
H
3
)
]
+
⇌
A
g
+
+
N
H
3
K
不稳1
Θ
[
A
g
(
N
H
3
)
2
]
+
⇌
[
A
g
(
N
H
3
)
]
+
+
N
H
3
K
不稳2
Θ
\begin{aligned} {\rm [Ag(NH_3)]^+} &\rightleftharpoons {\rm Ag^+ + NH_3 }&K_{\text {不稳1}}^\Theta \\ {\rm [Ag(NH_3)_2]^+} &\rightleftharpoons {\rm [Ag(NH_3)]^+ + NH_3} &K_{\text {不稳2}}^\Theta \end{aligned}
[Ag(NH3)]+[Ag(NH3)2]+⇌Ag++NH3⇌[Ag(NH3)]++NH3K不稳1ΘK不稳2Θ
第7章 电化学基础
原电池
原电池的符号表示
- 负极在左,正极在右,并用
(-)和(+)注明 - 溶液标明浓度,气体标明分压
- 用
|表示相界面,用||表示盐桥,用,分隔同相中的不同组分 - 固体、气体靠近电极,溶液靠近盐桥
- 无明显电极材料时添加惰性电极 P t \rm Pt Pt
- 本身未得失电子,但参与反应的组分( H + , C l − \rm H^+,Cl^- H+,Cl−等)应当写入相应的电极侧
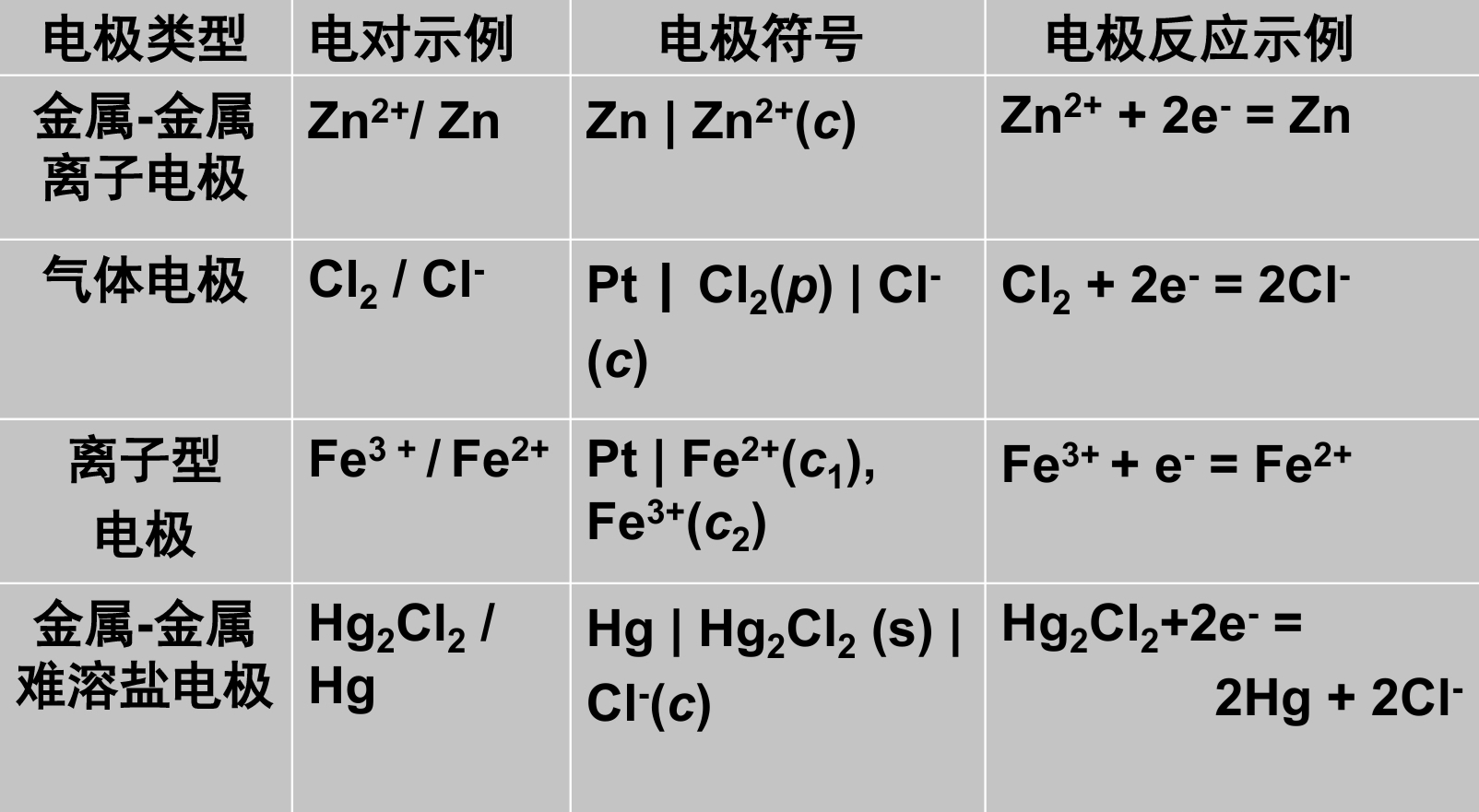
电极分类
电极电势
电极电势的产生:斯特恩双电层模型
电极电势 E ( M n + / M ) = E ( 金属 ) − E ( 溶液 ) E(\text M^{n+}/\text M) = E(金属) - E(溶液) E(Mn+/M)=E(金属)−E(溶液)
标准电极电势
参与电极反应的各物质均处于标准状态时的电极电势规定为标准电极电势 E Θ E^\Theta EΘ
规定 298 K 298 \;\text K 298K 下,标准氢电极 H + ( 1 m o l / L ) ∣ H 2 ( 100 k P a ) ∣ P t \rm H^+(1 \; mol/L) \;|\; H_2(100kPa) \;|\; Pt H+(1mol/L)∣H2(100kPa)∣Pt 的电极电势为 E Θ ( H + / H 2 ) = 0 V E^\Theta(\text H^+/\text H_2)=0 \;\text V EΘ(H+/H2)=0V
电极电势大,电对中氧化态物质氧化性强,易作正极
电极电势小,电对中还原态物质还原性强,易作负极
原电池热力学
电动势与吉布斯函数变
仅考虑可逆电池:
Δ
r
G
m
,
T
Θ
=
W
=
−
n
F
E
Θ
\boxed{ \Delta_r G_{m,T}^\Theta = W = -nFE^\Theta }
ΔrGm,TΘ=W=−nFEΘ
其中法拉第常量 F = 96500 C/mol F = 96500 \;\text{C/mol} F=96500C/mol
例题:
若两个原电池反应加和得到第三个原电池反应
则其电极电势运算关系:
C
l
2
+
2
e
−
=
2
C
l
−
E
1
Θ
=
+
1.36
V
C
l
O
3
−
+
6
H
+
+
6
e
−
=
C
l
−
+
3
H
2
O
E
2
Θ
=
+
1.45
V
C
l
O
3
−
+
6
H
+
+
5
e
−
=
1
/
2
C
l
−
+
3
H
2
O
E
3
Θ
\begin{aligned} \rm Cl_2+2e^- &= \rm 2Cl^- & E_1^\Theta =+1.36 \;\text V \\ \rm ClO_3^- + 6H^+ + 6e^- &= \rm Cl^- + 3H_2O & E_2^\Theta = +1.45 \; \text V \\ \rm ClO_3^-+6H^+ +5e^- &= \rm 1/2\; Cl^-+3H_2O &E_3^\Theta\quad\quad\quad\quad\quad \end{aligned}
Cl2+2e−ClO3−+6H++6e−ClO3−+6H++5e−=2Cl−=Cl−+3H2O=1/2Cl−+3H2OE1Θ=+1.36VE2Θ=+1.45VE3Θ
则
−
0.5
×
①
+
②
=
③
-0.5\times ①+② = ③
−0.5×①+②=③
反应一乘
0.5
0.5
0.5,此时标准电极电势仍然为
E
1
Θ
=
+
1.36
V
E_1^\Theta =+1.36 \;\text V
E1Θ=+1.36V
记一半的反应一的吉布斯函数变为
Δ
r
G
m
,
1
Θ
\Delta_r G_{m,1}^\Theta
ΔrGm,1Θ
∵
−
Δ
r
G
m
,
1
+
Δ
r
G
m
,
2
=
Δ
r
G
m
,
3
∴
n
1
F
E
1
Θ
−
n
2
F
E
2
Θ
=
−
n
3
F
E
3
Θ
∴
E
3
Θ
=
−
n
1
E
1
Θ
+
n
2
E
2
Θ
n
3
=
1.47
eV
\begin{aligned} &{\color{maroon}\because -\Delta_r G_{m,1} + \Delta_r G_{m,2} = \Delta_r G_{m,3} }\\ &{\color{maroon}\therefore n_1FE_1^\Theta -n_2FE_2^\Theta =-n_3FE_3^\Theta }\\ &{\color{black}\therefore E_3^\Theta = \dfrac{-n_1E_1^\Theta + n_2 E_2^\Theta}{n_3} = 1.47 \;\text {eV} } \end{aligned}
∵−ΔrGm,1+ΔrGm,2=ΔrGm,3∴n1FE1Θ−n2FE2Θ=−n3FE3Θ∴E3Θ=n3−n1E1Θ+n2E2Θ=1.47eV
其中 n 1 = 0.5 × 2 = 1 n_1 = 0.5 \times 2 =1 n1=0.5×2=1
电动势与平衡常数
由于
−
R
T
ln
K
Θ
=
Δ
r
G
m
Θ
=
−
n
F
E
Θ
\boxed{ -RT \ln{K^\Theta} = \Delta_rG_m^\Theta = -nFE^\Theta }
−RTlnKΘ=ΔrGmΘ=−nFEΘ
可知:
E
Θ
=
R
T
n
F
ln
K
Θ
=
0.0592
n
lg
K
Θ
E^\Theta = \frac{RT}{nF}\ln{K^\Theta} = \frac {0.0592} n \lg{K^\Theta}
EΘ=nFRTlnKΘ=n0.0592lgKΘ
非标态下的电极电势
对于任意电极反应,其非标态下的电极电势(能斯特方程):
E
=
E
Θ
+
R
T
n
F
ln
[
O
x
]
p
[
R
e
d
]
q
\boxed{ E = E^\Theta + \frac{RT}{nF}\ln{\frac{\left[Ox\right]^p}{\left[Red\right]^q}} }
E=EΘ+nFRTln[Red]q[Ox]p
若电极反应中还有 H + , O H − \rm H^+,OH^- H+,OH− 等其它物质参与,也应写入能斯特方程
溶液pH对电极电势的影响
有
H
+
,
O
H
−
\rm H^+,OH^-
H+,OH− 参与的电极反应,电极电势受酸度影响大
无
H
+
,
O
H
−
\rm H^+,OH^-
H+,OH− 参与的电极反应,电极电势不受酸度影响
沉淀与配合反应对电极电势的影响
转化为对浓度的影响
电解
分解电压与超电压
理论分解电压:克服电解产物所形成原电池产生的反向电动势所需外加电压
实际分解电压:保证电解能够连续进行的最低外加电压,简称分解电压
超电压:实际分解电压 - 理论分解电压。由电极极化产生(浓差极化、电化学极化)
电解产物的判断
阳极:电极电势小的电对先放电
若为非惰性金属电极,则电极放电。
一般放电顺序:
S
2
−
,
I
−
,
B
r
−
,
C
l
−
,
O
H
−
\rm S^{2-},I^-,Br^-,Cl^-,OH^-
S2−,I−,Br−,Cl−,OH−
阴极:电极电势大的电对先放电
电解盐溶液时,
E
Θ
E^\Theta
EΘ 大于或略小于
0
0
0 时(
Z
n
,
F
e
\rm Zn,Fe
Zn,Fe)金属离子放电,否则
H
+
\rm H^+
H+放电
电化学腐蚀
腐蚀电池
宏观电池:具有宏观可辨的正负极
微观电池:无明显分立的正负极
腐蚀速度快、程度重
腐蚀类型(依据产物分类)
析氢腐蚀:偏酸性环境
吸氧腐蚀
差异充气腐蚀(浓差腐蚀)
由氧气在金属表面分布不均匀形成
氧含量高处为正极
氧含量低处为负极,被腐蚀
金属防腐蚀
更换材料
保护层(涂层、磷化处理、氧化处理)
无机缓蚀剂(形成难溶盐薄膜)
有机缓蚀剂(形成难溶难透的有机薄膜)
牺牲阳极的阴极保护法
外加电流法
第8章 化学反应速率
化学反应速率的表示方法
a A ( g ) = b B ( g ) \rm {\it a}A(g) \xlongequal{\quad} {\it b}B(g) aA(g)bB(g)
反应进度
ξ
\xi
ξ
ξ
=
−
n
A
′
−
n
A
a
=
n
B
′
−
n
B
b
\xi = -\dfrac{n_A' - n_A}{a} =\dfrac{n_B' - n_B}{b}
ξ=−anA′−nA=bnB′−nB
反应速率定义1(
m
o
l
/
s
\rm mol/s
mol/s)
v
1
=
d
ξ
d
t
v_1 = \dfrac{\text d\xi}{\text dt}
v1=dtdξ
反应速率定义2(
m
o
l
⋅
L
−
1
⋅
s
−
1
\rm mol \cdot L^{-1} \cdot s^{-1}
mol⋅L−1⋅s−1)
v
=
d
ξ
V
d
t
v = \dfrac{\text d\xi}{V \text dt}
v=Vdtdξ
基元反应
基元反应:反应物一步转化为生成物的反应
复杂反应:反应物分步转化为生成物的反应
绝大多数反应为复杂反应
复杂反应到速率取决于最慢的一步
自由碰撞理论
(主要用于气相双分子)
发生有效碰撞的条件
足够的能量
合适的方向
对于基元反应:
m
X
(
g
)
+
n
Y
(
g
)
=
c
C
(
g
)
\rm {\it m}X(g) + {\it n}Y(g)\xlongequal{\quad} {\it c}C(g)
mX(g)+nY(g)cC(g)
| 有效碰撞频率决定因素 | 表达式 |
|---|---|
| 浓度决定碰撞频率 | Z = Z 0 ⋅ c m ( X ) ⋅ c n ( Y ) Z = Z_0 \cdot c^m(\text X) \cdot c^n(\text Y) Z=Z0⋅cm(X)⋅cn(Y) |
| 能量大于 E a E_a Ea 的比例 | f = e − E a / R T f = e^{-E_a/RT} f=e−Ea/RT |
| 碰撞角度合适的比例 | P P P |
因此,反应速率
v
=
P
f
Z
=
A
e
−
E
a
/
R
T
⋅
c
m
(
X
)
⋅
c
n
(
Y
)
\boxed{ v = PfZ = A e^{-E_a/RT} \cdot c^m(\text X) \cdot c^n(\text Y) }
v=PfZ=Ae−Ea/RT⋅cm(X)⋅cn(Y)
其中:
A
A
A 为与分子结构有关的常量
E
a
E_a
Ea 为反应活化能,等于有效碰撞所需最低分子动能与分子平均动能之差
浓度对化学反应速率的影响
质量作用定律
定义速率常数
k
k
k
k
=
A
e
−
E
a
/
R
T
k = A e^{-E_a/RT}
k=Ae−Ea/RT
则化学反应速率可表示为
v
=
k
⋅
c
m
(
X
)
⋅
c
n
(
Y
)
v = k \cdot c^m(\text X) \cdot c^n(\text Y)
v=k⋅cm(X)⋅cn(Y)
k k k 为速率常数,只与温度与催化剂相关
反应级数
反应分子数:对于基元反应,直接相互作用时参与的反应物分子数成为反应分子数
反应级数:
对于
v
=
k
⋅
c
m
(
X
)
⋅
c
n
(
Y
)
v = k \cdot c^m(\text X) \cdot c^n(\text Y)
v=k⋅cm(X)⋅cn(Y)
m
,
n
m,n
m,n 称为对应反应物的分级数
m
+
n
m+n
m+n 称为反应级数
对于基元反应,反应级数一般等于反应分子数
对于复杂反应,反应级数可以不等于反应分子数,且可以不为整数
零级反应
m X ( g ) = P r o d u c t \rm {\it m}X(g)\xlongequal{\quad} Product mX(g)Product
v = − d c t ( X ) / m d t = k ⇒ c t ( X ) = c 0 ( X ) − m k t v= -\dfrac{\text dc_t(\text X)/m}{ \text dt} = k \quad\Rightarrow\quad c_t(\text X) = c_0(\text X) - mkt v=−dtdct(X)/m=k⇒ct(X)=c0(X)−mkt
速率常数 k k k 的量纲: m o l ⋅ L − 1 ⋅ s − 1 \rm mol \cdot L^{-1} \cdot s^{-1} mol⋅L−1⋅s−1
一级反应
m X ( g ) = P r o d u c t \rm {\it m}X(g)\xlongequal{\quad} Product mX(g)Product
v = − d c t ( X ) / m d t = k c t ( X ) ⇒ { ln c t ( X ) − ln c 0 ( X ) = − m k t c t ( X ) = c 0 ( X ) ⋅ e − m k t \begin{aligned} &v= -\dfrac{\text dc_t(\text X)/m}{ \text dt} = kc_t(X) \\ \Rightarrow\quad & \!\!\! \begin{cases} \ln{c_t(\text X)} - \ln{c_0(\text X)} = -mkt \\ c_t(\text X) = c_0(\text X) \cdot e^{-mkt} \end{cases} \end{aligned} ⇒v=−dtdct(X)/m=kct(X){lnct(X)−lnc0(X)=−mktct(X)=c0(X)⋅e−mkt
速率常数 k k k 的量纲: s − 1 \rm s^{-1} s−1
存在半衰期 t 1 / 2 = ln 2 m k t_{1/2} = \dfrac{\ln{2}}{mk} t1/2=mkln2
二级反应
m X ( g ) = P r o d u c t \rm {\it m}X(g)\xlongequal{\quad} Product mX(g)Product
v
=
−
d
c
t
(
X
)
/
m
d
t
=
k
c
t
2
(
X
)
⇒
1
c
t
(
X
)
−
1
c
0
(
X
)
=
m
k
t
\begin{aligned} &v= -\dfrac{\text dc_t(\text X)/m}{ \text dt} = kc_t^2(X) \\ \Rightarrow\quad & \dfrac 1 {c_t(\text X)} - \dfrac 1 {c_0(\text X)} = mkt \end{aligned}
⇒v=−dtdct(X)/m=kct2(X)ct(X)1−c0(X)1=mkt
速率常数
k
k
k 的量纲:
m
o
l
−
1
⋅
L
⋅
s
−
1
\rm mol^{-1} \cdot L \cdot s^{-1}
mol−1⋅L⋅s−1
温度对化学反应速率的影响
阿伦尼乌斯公式
{
k
=
A
⋅
e
−
E
a
/
R
T
ln
k
=
−
E
a
R
⋅
1
T
+
ln
A
\begin{cases} k = A \cdot e^{-E_a/RT} \\ \ln k = \dfrac{-E_a}{R}\cdot \dfrac 1 T + \ln A \end{cases}
⎩
⎨
⎧k=A⋅e−Ea/RTlnk=R−Ea⋅T1+lnA
温度变化时,活化能越大的反应,速率变化越剧烈
低温下,反应速率对温度变化更敏感
催化剂对化学反应速率对影响
过渡态理论
考虑到碰撞中的化学键重排和能量重新分配
A
+
B
C
→
A
⋯
B
⋯
C
→
A
B
+
C
A+BC \quad\rightarrow\quad A \cdots B \cdots C \quad \rightarrow\quad AB + C
A+BC→A⋯B⋯C→AB+C
催化剂
- 只能对热力学允许的过程起作用
- 改变反应路径,降低活化能,从而改变反应速率
- 自身质量、化学组成不变(物理性质常常改变)
- 正逆反应活化能改变量相同,不改变平衡状态
- 特异性
- 会失活


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









