









人脑疾病多组学数据资源综述及综合分析
- Abstract
- Introduction
- Global burden of neuropsychiatric disorders and research efforts(神经精神疾病的全球负担和研究工作)
- Multi-omics and its use in research of neuropsychiatric disorders(多组学及其在神经精神疾病研究中的应用)
- Multi-omics data resources for neuropsychiatric disorders(神经精神疾病的多组学数据资源)
- Integrative multi-omics analyses in brain research(脑研究中的综合多组学分析)
- Limitations and future directions
Abstract
在过去的十年里,大量的组学数据集被用于人类大脑的研究。它的发展如此之快,迫切需要及时更新。本文综述了健康对照和神经精神疾病的主要多组学数据资源,包括精神分裂症、自闭症、双相情感障碍、阿尔茨海默病、帕金森病、进行性核上性麻痹等。综述了单细胞组学在脑研究中的最新进展,如单核RNA-seq、单细胞ATAC-seq和空间转录组学。我们进一步研究了组织和单细胞数据的综合多组学分析方法。最后,我们讨论了人类大脑疾病多组学研究的局限性和未来发展方向。

Introduction
Global burden of neuropsychiatric disorders and research efforts(神经精神疾病的全球负担和研究工作)
根据最新的全球疾病负担(GBD)研究的数据,在过去30年中,每10万人口中由神经精神疾病引起的死亡率增加了76%(图1A)。残疾调整生命年(DALYs;残疾生活年数和丧失生命年数的总和)也显示出不同神经精神疾病年龄组的显著差异(图1B)。例如,头痛和抑郁症在中青年中最为常见,而阿尔茨海默病和帕金森病等神经退行性疾病在75岁至95岁以上年龄组中最为严重。阿尔茨海默病和其他痴呆症在其他神经精神疾病中增加最多,部分原因是中国和美国等许多国家的人口结构老龄化。在美国,仅在2020年,65岁及以上痴呆症患者的医疗保健、长期护理和临终关怀服务的总支出估计就将达到3050亿美元。
了解人类大脑是最终找到治疗神经精神疾病的关键步骤之一。在过去的几十年里,许多国家已经认识到这一迫切需要,并启动了大型项目,包括美国的大脑计划、欧盟的人类大脑计划、中国的大脑计划、加拿大的大脑研究战略、澳大利亚的大脑联盟、日本大脑/MINDS项目和最近启动的国际大脑倡议。虽然这些大脑计划可能有不同的目标和不同的重点,例如,欧盟人类大脑计划最初的目标是建立一个大脑刺激器,但它们现在有一个共同的组成部分——使用最新的组学技术来了解脑细胞的分子功能及其在神经精神疾病中的作用。
(https://pubmed.ncbi.nlm.nih.gov/32135092/)
Multi-omics and its use in research of neuropsychiatric disorders(多组学及其在神经精神疾病研究中的应用)
两种主要的研究类型通常用于研究人类神经精神疾病:遗传研究和生物标志物研究。这项基因研究是基于大多数神经精神疾病都有遗传根源这一事实。遗传学研究可以检测与疾病相关的常见单核苷酸多态性(snp),受影响后代的新生变异和患者体内富集的罕见拷贝数变异。虽然这些遗传发现对疾病的机制提供了有限的认识,但遗传研究中的主要问题包括:(1)基因功能的不完整注释;(2)因果变量映射不精确;(3)关联与因果之间的差距;(4)对基因组非编码部分遗传变异的作用认识不足。另一种更以翻译为导向的方法是发现生物标志物。生物标志物研究针对不同状态患者的生物学变化。在比较患者与对照组,治疗与drug-naïve患者,或反应者与无反应者时,已经探索了各种生物材料。基于生物标志物的疾病分型和患者亚组可能对优化治疗有价值。
遗传和生物标志物研究在很大程度上依赖于多组学数据来实现其目标。多组学数据是指多个“组”的全系统数据,如基因组、甲基组、转录组、表观基因组、蛋白质组、代谢组和微生物组。多组学数据经常用于标注疾病相关的snp和基因组区域,构建假定的调控网络,并评估潜在的因果或中介关系。多组学可以协同解释神经精神疾病的生物学机制。多组学数据为机制发现的独立生物学验证提供了独特的支持。遵循中心法则,多组学数据可以帮助我们追踪与遗传变异、环境变化和受影响状态相关的不同生物学水平的分子变化。
在过去的10年里,产生了大量的多组学数据。其中一些特别存在于大脑中,这对神经精神疾病的研究至关重要。其他非脑数据也有参考价值(如宫内环境对精神分裂症风险的影响)。许多多组学数据是在患者和对照组的匹配组织中产生的,而其余数据仅来自健康对照组。其中一些数据来自少数捐赠者的组织或少量培养的细胞,而其他数据来自人群队列。从相当大的样本中可以得出种群变异,这对遗传研究很重要。除了每个组学的原始定量数据外,通常还使用参考图谱、定量性状位点(QTL)等衍生物。它们是遗传研究和生物标志物发现的重要资源。本文综述了可用于神经精神疾病研究的多组学数据的主要来源以及相关工具。
在这篇综述中,我们关注的是来自健康对照和神经精神疾病患者的人脑样本的组学数据。组学包括测量基因组DNA的丰度和变异,各种DNA甲基化,编码mRNA和非编码RNA (microRNA,长链非编码RNA或lncRNA),通过核糖核酸序列和蛋白质测量的主动翻译RNA,以及其他表观基因组特征,包括通过ATAC-seq或DNA -seq测量的染色质可及性,Hi-C三维染色体和基因组结构。大部分数据是通过下一代测序产生的。少量数据来自微阵列或Illumina BeadChip。这些不同的组学数据涵盖了中心教条的主要生物成分。不同程度覆盖了不同的脑区和发育阶段。单细胞数据被单独描述,因为它捕获了细胞类型特异性的特征,而这些特征可能被大块组织数据遗漏。
由于篇幅限制,我们没有涵盖许多其他对研究神经精神疾病有用的表型数据。**例子包括脑成像数据、生理和行为特征以及动物模型 **。所有这些都可以为疾病机制建模和制定预测算法提供信息。
Multi-omics data resources for neuropsychiatric disorders(神经精神疾病的多组学数据资源)
我们调查了产生一般人类生物学或特定神经精神疾病多组学数据的主要队列或项目(见表1中的摘要)。我们必须强调,下面列出的联盟列表可能不完整。由于篇幅限制,这里没有列出许多其他重要的组学资源。我们也可能会错过其他神经精神疾病的一些个体组学数据集,例如,自闭症大脑皮层[7]的24个总RNA-seq,多发性硬化症[8]和>的15个大脑的RNA-seq,癫痫[9]的110 K单细胞神经元转录组。这个领域正在快速发展。来追踪最新的人类大脑多组学数据集,我们保持数据源表[10]公开可编辑,以便每个人都可以为列表做出贡献。
General consortia 综合性队列研究

DNA元件百科全书(ENCODE)项目最初于2003年启动,是一项国际努力,旨在描述大约1%的人类基因组的调控功能。进一步的开发包括收集基于测序技术的全基因组数据,如RNA-seq、DNA甲基化、染色质免疫沉淀测序(组蛋白和TF的ChIP-seq)、DNA酶I超敏位点,这些分析在多个细胞系[12]中进行。转录因子结合位点数据是一种独特的资源,不容易从其他项目[13]中获得。近年来,数据类型的扩展,如染色质相互作用的配对末端标记分析(china - pet),高通量染色质构象捕获技术(Hi-C)。截至2020年12月,ENCODE实验矩阵显示了10 314个人类多组学数据集。尽管ENCODE主要关注细胞系,但最近的更新包括一些来自人脑、原代神经元或神经元细胞系的组学数据(即截至2020年12月,在人脑器官进行的449次实验)。在目前的第4阶段,ENCODE项目继续进行人类基因组的数据汇编、计算分析和功能表征。
Roadmap表观基因组学项目收集了主要来自人类血液和22种组织类型的RNA-seq、ChIP-seq(组蛋白)、dna -seq和甲基化数据。尽管该项目于2018年结束,但开放获取的表观基因组数据(现在包含在ENCODE数据门户网站中)仍然是多组学整合分析的可行资源。截至2020年12月,ENCODE实验矩阵显示了路线图中发布的2154个多组学人类数据集,其中包括158个与成人和胎儿大脑相关的数据集。
在过去的20年里,哺乳动物基因组功能注释(Functional AnnoTation Of the哺乳动物基因组,FANTOM)联盟收集了大量资源来了解健康人类/小鼠细胞和组织的转录调控,包括非编码转录物。基因表达的Cap分析(CAGE)数据是该项目非常独特的数据,对于定义转录起始位点至关重要。FANTOM5创建了最全面的增强子和启动子目录,其中包括脑特异性注释。FANTOM6专注于非编码RNA的注释,这是目前基因组注释的主要弱点。FANTOM数据已用于许多多组学工具,如HACER (Human Active enhancer to interpret Regulatory variants),该工具整合了FANTOM5、表达数量性状位点(eQTL)数据库、ENCODE转录因子结合位点(TFBSs)、新生RNA测序(GRO/PRO-seq)和high - c数据。包括脑细胞特异性分析。
基因型-组织表达(GTEx)项目旨在描述个体和人体不同组织中基因表达水平的变化。除了发现顺式eQTL外,还发现了组织特异性的反式eQTL,以及跨细胞类型的eQTL相互作用,性染色体特异性的eQTL。处理后的数据可供下载,并可通过web界面访问。值得注意的是,GTEx具有多个大脑区域的转录组谱,样本量相当大(n<255)。
Omics Discovery Index (OmicsDI)是一个搜索多组学数据集的平台。它集成了来自多个数据库的蛋白质组学、基因组学、代谢组学和转录组学数据集,从GEO和欧洲基因组-表型档案(EGA)到集成网络细胞特征库(LINCS)、dbGaP等。它可以通过生物体、疾病、组织、基因标识符和关键字搜索数据。截至2020年12月,对“大脑”的搜索产生了116 261个结果,其中10个是多组学数据集。
Brain-specific consortia
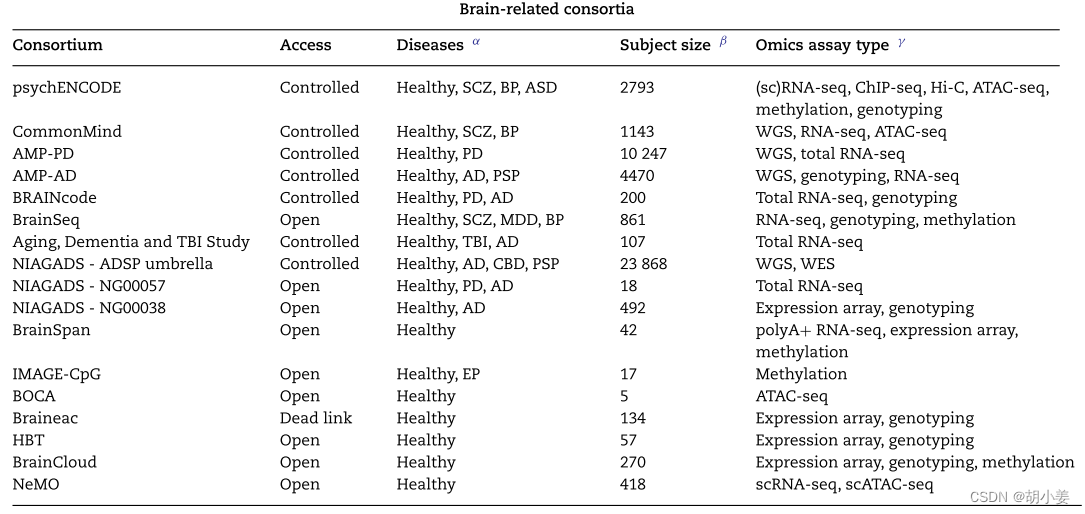
艾伦脑科学研究所(Allen Institute for Brain Science)是最早致力于创建与解剖结构相关的脑特异性组学数据系统资源的机构之一。Allen人脑图谱收集了来自两个个体的大约900个大脑神经解剖切片的微阵列表达谱和MRI测量,有效地证明了基因表达与空间定位[25]相关。进一步的发展包括收集人类和小鼠大脑发育的时空基因表达程序,衰老,痴呆,创伤性脑损伤(TBI)和IVY胶质母细胞瘤图谱项目。两个这样的项目包括BrainSpan(对42个发育中的人类大脑的16个大脑区域进行rna测序)和老化、痴呆和TBI研究(107个受试者的总rna测序)。最新增加的包括单细胞RNA测序和原位杂交,来自人类、小鼠和恒河猴皮层、海马体、脊髓的各个部分,以及丘脑的比较细胞解剖。Patch-seq是另一项新技术。数据可在线免费获取或通过编程访问(AllenSDK,大脑建模工具包,DiPDE仿真平台)。虽然基因表达是艾伦脑图谱的主要焦点,但电生理和形态学数据的可用性使其成为脑空间转录组学的独特资源。
PsychENCODE联盟成立于2015年,专注于人类大脑的基因组学和表观基因组学数据,用于研究神经精神疾病b[27]。PsychENCODE数据的特点是最大的大脑集合(2793个独特的捐赠者)的对照和主要精神疾病,包括精神分裂症,双相情感障碍和自闭症。大多数数据来自出生后的成人大脑。但是发育方面也一直被用于研究发育障碍[28,29]。几乎所有的大脑都有基因型数据,这使它们成为绘制分子定量性状位点的最有力数据。第一次发布的数据主要来自大块组织。RNA-seq转录组可用于所有组织。组蛋白标记ChIP-seq, ATAC-seq,核糖核酸-seq,蛋白质组学,DNA甲基化,Hi-C数据可用于某些组织。额叶皮质是该联盟研究的主要脑区。对第一次发布的部分数据的分析提供了11篇研究论文。
除了可控制访问的原始数据外,psychENCODE还提供了一个不断增长的经过处理的、衍生的和综合的结果列表,如大脑表达基因列表、疾病相关基因、共表达模块、大脑单细胞表达谱、组织修饰数据、超过79个kenenh子、染色质环、和拓扑相关结构域,超过2.5 M的eqtl和snp与剪接、细胞特异性和染色质活性相关。将转录因子和增强子与靶基因连接,可以创建321个基因网络。这些数据已被用于训练机器和深度学习模型,以预测精神病表型,其性能比加性多基因风险评分[31]提高了6倍以上。作为ENCODE、Roadmap和brain Atlas数据的补充,PsychENCODE数据是评估人脑组学群体变异的最佳数据之一。GTEx因其多组织eQTL而广受欢迎,但由于其规模和全面性,与来自PsychENCODE的脑特异性数据相比,GTEx相形见绌。PsychENCODE正在进入第二阶段,重点是单细胞数据。
加速药物伙伴关系(AMP)是NIH、制药公司和非营利组织之间的竞争前伙伴关系,专注于确定和验证诊断和药物开发的有前途的生物靶点。AMP- ad是AMP框架下的三个初始项目之一,预算为5年1.852亿美元。AMP-AD的目标是应用尖端的系统和网络生物学方法,将来自2000多个阿尔茨海默病(AD)各个阶段的人类大脑的多维人类分子数据(基因组、表观基因组、RNA、蛋白质组学)与临床和病理数据整合在一起。贡献最多多组学数据的三个最大的AMP-AD研究是ROSMAP、MSBB和MayoRNAseq。例如,西奈山脑库(MSBB)的研究为来自四个大脑区域(颞上回、额极、海马旁回)的1700多个大脑样本提供了总RNA测序额下回)。大多数受试者还进行了全外显子组测序(WES)、全基因组测序(WGS)和蛋白质组学质谱分析。除了多组学数据外,还为大多数参与者提供了全面的临床和神经病理学评估,包括Braak ad分期评分、神经斑块密度、临床痴呆评定量表(CDR)、最小状态检查(MMSE)、CERAD评分[32]和组织学染色。另一项AMP-AD研究ROSMAP为多达2214个脑样本提供基因组学、转录组学、蛋白质组学、表观基因组学(例如H3K9ac ChIP-seq、DNA甲基化)和代谢组学。ROSMAP还生成了超过48个AD患者和无认知障碍受试者大脑的单细胞转录组学数据[33,34]。丰富的AMP-AD数据集与其他AD项目一起托管在AD知识门户网站(https://adkno wledgeportal.synapse.org)上,该网站是AD研究的最大数据门户之一。
AMP旗下针对神经疾病的另一个项目是AMP- pd (https://amp-pd.org)。AMPPD于2018年启动,旨在通过对现有队列中PD患者数据和生物样本的深入分子表征和纵向临床分析,识别和验证帕金森病(PD)的诊断、预后和/或疾病进展生物标志物。amp - pd的最初发布包括来自四个队列的4298名参与者:Michael J. Fox基金会(MJFF)和国家神经疾病和中风研究所(NINDS) BioFIND研究,哈佛生物标志物研究(HBS), NINDS帕金森病生物标志物计划(PDBP)和MJFF帕金森病进展标志物计划(PPMI)。其最新版本扩展到包括来自7个队列的10247名参与者,为PD和对照组提供临床、基因组学和纵向转录组学数据。请注意,该纵向项目在不同的就诊时间(如基线、0.5、6、12、18、24和36个月)收集全血样本进行总RNA测序。在AMP-PD网站上,共有8461个RNA-seq数据集(来自n=3274名参与者)以及统一的临床测量数据,为研究帕金森病的进展提供了一个很好的资源。AMP-PD程序将其数据托管在谷歌云平台(GCP)中,并建议用户通过Terra平台(http://app.terra.bio/)访问、运行分析工具和协作。考虑到像这样的大型组学队列将产生越来越多的数据的趋势,将代码和工具带入数据,而不是将数据下载到本地服务器,将是一个更实用的解决方案。
BRAINcode项目(http://www.humanbraincode.org)has)旨在通过对人类神经元基因组、转录组和表观基因组的整合分析,解码易受神经退行性疾病影响的大脑神经元的功能(如PD的多巴胺神经元,AD的锥体神经元)。在最初的发布中,它包括从大约100个人类死后大脑中激光捕获的50,000个神经元中超深核糖耗尽的总RNAseq,包括来自实体黑质(n=86个大脑)的多巴胺神经元和来自皮层(n= 10个大脑)的锥体神经元。在多巴胺神经元中发现了超过71000个新的转录非编码元件(TNEs),其中许多与活性增强子(oreRNAs)[36]的表观遗传特征一致。eQTL分析和GWAS共定位分析发现,这些eRNA元素在多巴胺低活性性状(如PD)和多巴胺多活性性状(如精神分裂症和成瘾)的遗传变异中都富集。BRAINcode的第二阶段包括来自AD和PD患者的100多个大脑,并在脑转录组中确定了bb1011000个神经元环状rna(数据将很快发布)。BRAINcode正在进入新的阶段,专注于对另外200个人类大脑进行单核RNA-seq、单核ATAC-seq、空间转录组和单细胞eQTL的研究。
CommonMind Consortium (CMC)生成了一个来自背外侧前额叶皮层(DLPFC;Brodmann区域9和46)对来自四个独立脑库的约1000人进行了研究,其中包括501名对照个体,353名诊断为精神分裂症的个体和120名诊断为双相情感障碍的个体。基因组数据包括980个个体的RNA-seq和SNP基因型,269个个体的ATAC-seq基因型,其中264个是RNA-seq个体的子集。原始数据和处理后的数据都可以在Synapse平台上获得,网址是http://CommonMi nd.org。
BrainSeq是由Lieber研究所和制药公司领导的一个项目,旨在描述主要神经精神疾病和对照样本中不同大脑区域转录组的遗传和表观遗传调控[38,39]。该联盟的第一阶段包括对738名受试者的DLPFC polyA+ RNA-seq,这些受试者跨越生命周期和三个主要的精神诊断组(精神分裂症、重度抑郁症和双相情感障碍)。二期扩展到海马体区域,并对551名跨越生命周期的受试者和成年精神分裂症患者进行了ribozero治疗的总rna测序。在BrainSeq的前两个阶段,总共有861位独特的捐赠者。还有第二阶段的Illumina 450 k芯片数据,用于分析海马和DLPFC的DNA甲基化[40,41]。他们还在http://eqtl.brainseq.org上提供了与发育和精神分裂症相关的基因图谱和eQTL资源。值得注意的是,利伯研究所还启动了非洲祖先神经科学研究计划,旨在向公众提供世界上最大的关于最近非洲祖先的人类死后大脑的组学数据集。
神经科学多组学档案(NeMO档案)是一个数据存储库,专门用于存储和传播由BRAIN计划和相关大脑研究项目产生的组学数据。NeMO数据包括人类、小鼠和狨猴的转录活性、甲基化、组蛋白修饰谱和染色质可及性。目前在公共BRAIN倡议细胞普查网络(BICCN)上对人类数据的搜索显示,418个样本具有开放获取的单细胞RNA-seq (n= 412)和单细胞ATAC-seq (n= 6)。更多的人类大脑单细胞组学数据(如单细胞place -seq, ATAC-seq, RNA-seq数据,用于定义细胞类型特异性的3D表观基因组[42])可以通过限制访问在NeMO上获得。
Integrative multi-omics analyses in brain research(脑研究中的综合多组学分析)
Multi-omics integration at single-cell level(单细胞水平的多组学整合)
大脑的高细胞复杂性促使单细胞组学方法在单细胞水平上理解基因组调控的应用。Darmanis等人率先提供了466个健康人皮质细胞的单细胞转录组学数据。随后的研究包括神经发生期间的时间过程scRNA-seq分析,揭示了谱系特异性轨迹和神经发生转录因子[44]的动态。单细胞甲基化数据也被用于揭示人类皮层[45]中的神经元亚群。最新的研究,如ROSMAP队列中48名阿尔茨海默病患者和健康个体的单细胞转录组学分析,已经扩大到80个K细胞,揭示了前所未有的疾病病理生理学的见解。最近,STAB,一个人类大脑的时空细胞图谱,通过分析13个可用的人类大脑scRNA-seq数据集[47],定义了跨越20个大脑区域和11个发育时期的42个细胞亚型。虽然人类单细胞数据仍然稀缺,但各种资源提供小鼠scRNA-seq数据,其中DropViz (http://www.dropviz.org/, 690 K细胞)和10X Genomics (1.3 M细胞[48])目前是最大的。
scRNA-seq技术已经发展到包含其他层的多组学信息,开放染色质是最近添加的技术之一。Lake等人的[50]将单核RNA测序(snDrop-seq)与单细胞开放染色质谱分析(scts -seq)结合在一起,对来自成人视觉皮层、额叶皮层和小脑的60000多个细胞进行了研究,显示出更好的细胞亚群分辨率和从另一个细胞中预测组学数据的能力。同样,Trevino等人在[51]中整合了人类前脑特异性RNAseq和ATAC-seq数据,揭示了详细的增强基因活性相关性,神经发生特异性转录因子的时间活性以及与神经精神疾病易感性相关的细胞类型和时间段。Li等人整合了来自psychENCODE和BrainSpan的scRNA-seq、小RNA-seq、组蛋白-seq和甲基化数据,概述了与神经精神疾病相关的人类大脑发育和细胞类型特异性基因表达模块的功能基因组学。有关这些和其他研究的详细信息可在https://github.com/mdozmoro v/scRNA-seq_notes#brain上获得。
通过整合来自基因分型阵列或WGS的遗传变异,scRNA-seq还允许我们在不同细胞类型和动态过程中绘制eqtl,其中许多在使用大组织方法时是模糊的。为了将这项技术应用于大规模的群体遗传学研究,Luke Franke和他的同事们建立了单细胞eQTLGen联盟(sc-eQTLGen),旨在精确定位致病基因变异影响基因表达的细胞环境。
空间转录组学的最新发展,如10X Genomics Visium[54,55]、Slide-seq[56]、HDST[57]、MERFISH[58]和LCM-seq[36,59],使定位特异性单细胞基因表达程序[60]得以明确鉴定。这些技术开始被应用于揭示人类DLPFC的分层结构,该结构由不同的基因表达程序[55]标记。重要的是,空间转录组学与其他数据(如神经精神基因集)的整合,证明了疾病相关信号[55]的位置特异性相关性,开启了整合多组学的新篇章。更多脑特异性单细胞空间转录组学数据集正在变得可用。其他一些方法使用scRNA-seq数据来解析空间表达
另一个新发现是脑细胞中的三维染色质组织。一些研究使用Hi-C及其变体将人类大脑的3D基因组学与基因表达、组蛋白修饰(ChIP-seq)、开放染色质(ATAC-seq)和gwas信号整合在一起,证明了基因组空间组织的重要性[5,64 - 66]。染色体构象捕获技术已扩展到单细胞水平,并与基因表达相结合,揭示了三维结构与基因表达之间的关联。最近的发展包括在单细胞中同时捕获染色质构象和甲基化的技术。
Methods and tools for multi-omics data integration(多组学数据集成的方法与工具)
正如Subramanian等人[71]所总结的那样,多组学数据集成的目标大致可以分为三类:1.多组学数据集成;基于多组学特征的疾病亚型和分类2. 生物标志物的各种应用预测;3. 获得生物学见解。例如,在疾病遗传学研究中,我们通常通过eQTL分析将基因组和转录组数据结合起来,以提高对具有功能影响的常见变异的检测(如GTEx研究[22])。然而,这种方法并不容易,因为环境因素和疾病状态也会影响转录组。Mohammadi等[72]开发了ANEVA-DOT,通过比较个体的母本和父本等位基因的表达活性来鉴定对基因表达影响异常强烈的杂合DNA变异。Montaner等人[73]最近回顾了多组学数据(包括蛋白质组学、基因组学、转录组学和代谢组学)的综合分析,并为中风发病机制、治疗靶点鉴定和生物标志物发现提供了有用的见解。实现这一目标的方法可分为早整合和晚整合两种,前者将组学矩阵组合成一个然后对其进行分析,后者分别分析每个组学模式然后将结果结合起来,参见文献[74]。或者,积分方法可以分类为无监督(矩阵分解,基于关联的,贝叶斯方法,基于网络的方法)或监督(基于网络的方法,多核学习),参见参考文献。[75-77],在Tini等人[78]中进行了基准测试,其中许多是在mixOmics R包中实现的[79]。最新的发展包括神经网络架构,如变分自编码器(VAE),用于数据集成[80]。作为这些疾病分类方法的应用,Zhang等[81]最近开发了一种端到端基于vae的模型OmiVAE,用于从多组学数据中提取低维特征并对样本进行分类。通过整合全基因组DNA甲基化和基因表达谱以及450804个分子特征,他们使用来自the Cancer Genome Atlas (TCGA)的泛癌症多组学数据集,对33种肿瘤类型和正常肿瘤的9081个样本进行了评估[81]。经10倍交叉验证,OmiVAE对33种肿瘤类型和正常样本的平均分类准确率为97.49%,优于现有的其他方法。
与大量组织数据相比,单细胞组学数据提供了高分辨率细胞亚型中高表达基因的更准确的转录组分析,但同时存在成本高、覆盖率低、深度浅和数据缺失率高等问题。因此,单细胞组学需要新的算法,同时采用用于批量组学数据分析的技术。我们提供一些最具代表性的单细胞集成方法的示例,并参考前面的评论以获得更详细的概述。整合单细胞组学数据的方法包括使用非负矩阵分解(NMF)[82]或类似的降维或低维嵌入方法[83]。
将scRNA-seq与其他单细胞数据整合的一个示例方法是LIGER,这是一种基于nnf的方法,用于整合和分析多个单细胞数据集,跨越条件、技术(scRNA-seq、甲基化、空间转录组学)或物种(人类和小鼠)[84]。当应用于人类和小鼠脑细胞时,它解决了其他不可观察的空间细胞状态。基于共享嵌入的方法Harmony已被用于整合scRNA-seq和空间转录组学数据[85]。在Seurat R包的Signac扩展中,已经实现了使用潜在语义索引(LSI)和改进的术语频率逆文档频率(TF-IDF)过程集成scRNA-seq和scATAC-seq[86]。通过降维和聚类,ArchR R包在整合scATAC-sec和scRNA-seq数据方面优于Signac[87]。基于网络相似性的CellWalker方法已被证明对scRNA-seq和scATAC-seq数据的稀疏性和噪声具有更强的鲁棒性[88]。它已被应用于发育中的人类大脑,并鉴定了在转录状态之间转换的细胞,解析了细胞特异性增强子,并通过增强子将神经特征相关基因定位到特定的细胞类型[88]。MAESTRO工具套件利用综合数据分析的最佳实践(例如基于图和基于密度的聚类,从染色质可及性建模基因调控潜力)全面整合scRNA-seq和scATAC-seq数据,并为注释细胞簇提供血液和大脑特异性细胞签名[89]。三维基因组整合方法也开始出现。最近提出了一种基于nmf的方法,使用scRNA-seq和scATAC-seq对大量Hi-ChIP信号进行亚群体特异性反卷积[90]。这些方法展示了单细胞多组学数据整合的潜力,揭示了复杂细胞系统(如大脑和神经精神疾病)的新生物学见解。
A case study of schizophrenia with multi-omics data
作为分析组学数据的案例研究,我们希望通过整合PsychENCODE和其他联盟[31]中的多组学数据来强调精神分裂症的研究。整合分析[31]将来自PsychENCODE、GTEx、ENCODE、CommonMind、Roadmap表观基因组学和单细胞分析的多组学数据合并到一个金字塔结构中,例如原始数据文件的基础;中间层是统一处理和易于共享的结果(如开放染色质区域和基因表达量化),顶层是基于调控网络和qtl的综合深度学习模型的“上限”。
**·**在转录组学水平上,他们使用基于NMF的方法对大组织RNA-seq数据进行反卷积,并将NMF顶部成分(NMF- tc)与单细胞衍生的细胞类型特征进行比较。在验证后,他们然后用单细胞特征对大组织RNA-seq进行反卷积,以估计个体间的细胞分数。他们表明,单细胞特征的加权组合可以解释大块组织中大多数群体水平的表达变化,准确率为0.88%。然后,他们比较了各种神经精神特征中每种细胞类型的细胞分数变化,并报告了疾病特异性细胞比例变化(见[31]中的图S15)。
**·**在表观基因组学水平上,他们通过整合PsychENCODE中的H3K27ac和H3K4me4 ChIPseq数据以及来自Roadmap PFC样本的dna -seq和ChIP-seq数据,在PFC中注释了79 056个增强子。然后,他们通过执行表达、剪接异构体、染色质和细胞分数QTLs(分别为eQTLs、isoQTLs、cQTLs和fQTLs)来鉴定影响基因表达和染色质活性的QTLs。他们在PFC中发现了33000个eGenes,接近估计在大脑中表达的基因总数,反映了他们的大样本量。他们还测试了不同基因组注释中各种qtl的富集程度。它们在eqtl和cqtl之间表现出最大的交集。他们发现了2477个多qtl。通过整合Hi-C相互作用数据,他们发现,与直接位于eGene启动子或外显子的SNPs相比,涉及远端eGenes但由Hi-C相互作用连接的qtl表现出明显更强的相关性。
**·**他们通过连接增强子、tf和靶基因进一步构建了基因调控网络。例如,他们使用弹性网络回归的系数(例如,假设目标基因的表达是由其调节TF的表达水平的线性组合决定的)来推断TF与目标基因之间的联系。基于调控网络,他们进一步将非编码GWAS变体与疾病基因联系起来。他们确定了1111个假定的SCZ相关基因(SCZ基因),其中321个得到了两个以上证据来源的支持(例如qtl和Hi-C)。有趣的是,他们发现大多数SCZ基因甚至不在具有指数snp的LD中(~ 67%,r2 <0.6)。最后,他们将这些SCZ基因与单细胞图谱联系起来,发现它们在神经元中高度表达,尤其是兴奋性神经元。
**·**最后,为了充分捕捉基因型和表型之间的相互作用,他们结合了一个可解释的深度学习框架,即深度结构化表型网络(DSPN)。与逻辑回归等传统分类方法不同,DSPN可以包括具有稀疏连通性的分子表型(如基因表达、增强子、共表达模块、细胞分数)的中间层。DSPN能够在预测性状方面获得更大的6倍改进,这可能反映了其纳入非线性相互作用的能力。对于精神分裂症,完整DSPN模型解释的方差超过了普通snp解释的方差(例如32.8%对25.6%)。
Limitations and future directions
Newly generated data
理想情况下,我们将需要涵盖所有生物学水平的多组学数据,从DNA到蛋白质的中间步骤,从干细胞到死亡的所有发育阶段,从神经元到神经胶质细胞的所有细胞类型,以及从未使用药物到接受各种治疗的患者的所有状态。在现实中,只有一小部分所需的数据已经生成并准备好使用。除了以上总结的数据,我们知道在未来的1 - 2年内将会有更多的单细胞转录组、ATAC-seq数据。空间转录组、Hi-C数据将有助于更好地理解脑转录组及其调控。将产生越来越多的eQTL和其他分子QTL大脑,脑细胞在不同的发育阶段,不同的种族背景和性别。令人鼓舞的是,评估来自同一细胞的多组学信号的技术开始出现,例如评估scRNA-seq和scATAC-seq数据的SNARE-seq[91]。
一些特定的组学数据仍然未被充分代表。DNA甲基化数据,microRNA表达和蛋白质组学数据是可以更好地覆盖的例子。在Illumina停止生产该试剂盒后,核糖测序(或核糖体分析)数据不太可能增加。脑细胞中转录因子的ChIP-seq数据是一个主要的数据类别,不幸的是大部分缺失。线粒体相关基因组学和表观基因组学还没有得到足够的重视。
我们还应该注意到-omics对性别、种族、遗传背景等变量很敏感,并且在大多数公共数据库中效果并不好。种族多样性的数据仍然广泛缺乏。2019年,研究人员发现,约78%的GWAS个体具有欧洲血统[92]。大脑组学数据的多样性更少。例如,在当前的AMP-PD发布中,不到4%的参与者是非白人/高加索人。这对于想要更具包容性的研究来说是一个巨大的问题。像非洲血统神经科学研究这样的项目有望缩小这一差距。
The ‘true’ single-cell transcriptome for human brains
人脑研究中的大多数单细胞转录组数据实际上是单核RNA-seq,而不是单细胞RNA-seq,因为目前的技术难以在不破坏细胞膜的情况下从死后冷冻的脑组织中提取完整的神经元细胞。此外,除了少数单细胞总RNA测序的实验尝试(如SuPeR-seq[93]、MATQ-seq[94]、RamDA-seq[95]和DART-seq[96])外,目前大多数单细胞RNA测序研究都是基于富含多聚a的RNA测序方法,导致许多有趣的非多聚a RNA(如miRNAs、piRNAs、circRNAs、eRNAs)未被关注。单细胞多组学的集成受到单细胞数据中存在的一些问题的影响,如单细胞数据中的dropouts问题以及细胞簇定义和注释中的分辨率问题。这些也将是集成研究的巨大挑战。单细胞/集成分析工具的验证和基准测试也是迫切需要的。
Human brain specimens versus cell lines
研究神经系统疾病的典型方法涉及健康和患病个体的人脑标本。与广泛获取的血液标本相比,大脑标本通常是从死后组织中获得的,这有其局限性:组织降解是主要原因。RNA对人死后的时间特别敏感。另一个限制是,死后组织只能提供生物系统的快照,这可能不足以揭示症状和治疗反应的动态。培养细胞和新开发的脑类器官具有环境因素和细胞组成相对均匀的优势,是生成多组学数据的重要替代方法。
Host-microbiome multi-omics integration
除了宿主本身,与微生物组学数据的整合也是一个有趣的方向。由于肠道微生物群是炎症物质的重要激活剂,研究人员观察到肠道中梭状芽胞杆菌等免疫调节微生物群的表达增加导致应激易感大鼠大脑中小胶质细胞密度和IL-1β表达增加[97]。最近对阿尔茨海默氏症的研究表明,肠道感染可能引发阿尔茨海默氏症大脑中淀粉样蛋白团块的产生(见综述[98])。虽然这些肠脑关联背后的机制在很大程度上尚不清楚,但宿主和微生物之间的多组学整合可能会带来新的见解[99,100]。
Longitudinal multi-omics analysis
大多数人类神经精神疾病的多组学分析是横断面的(例如,病例与对照,疾病亚型)。将组学分析纵向结合临床措施和治疗结果,可以提供更全面的评估,以提高疾病风险预测、早期发现和更好的治疗。以前的纵向多组学研究已经成功地确定了少数疾病的疾病标志物[101-103],但在神经精神疾病中却不多。我们在这里回顾的多组学队列之一AMP-PD包括纵向血液RNA-seq数据和帕金森患者的临床数据。我们期待在神经精神病学研究中获得更多的纵向组学数据。这种纵向数据通常来自外周组织。因此,需要对大脑和外周组织进行多组学比较分析来验证大脑的相关性。
Correlation versus causality analysis
许多多组学研究产生了相关结果。例如,eQTL分析是为了确定遗传变异与基因表达之间的相关性。许多所谓的“生物标志物”实际上是与疾病、特征或状态相关的生物分子信号。我们知道相关性并不能证明因果关系。一个典型的例子是GWAS的先导变异不一定是性状/疾病的致病变异[104]。已经开发了几种统计精细映射方法来提示GWAS输出的潜在因果关系[105]。机器学习和深度学习已被用于发现多组学数据中的模式和相关性,这在许多情况下(例如肿瘤识别、疾病预测)可能足够有效。然而,如果一个模型能够捕捉到因果关系,那么它将更加一般化。此外,如果我们能从结果中找出原因,我们就能更好地找到治疗疾病的方法。一些互补的方法(如孟德尔随机化、结构方程建模、贝叶斯网络)已被应用于发现基因组和表观基因组变异对脂质表型的新因果效应
High-dimension reduction challenge
多组学集成的另一个挑战是高维。尽管我们在这里回顾的许多多组学队列提供了大样本量,但样本量仍然远远少于特征数量(n p)。随着受试者被评估的特征越来越多,这正在成为一种趋势(例如,UK Biobank有50万参与者,每个参与者都有9600万个snp)。数千种临床/生活方式表型和大约4000种影像学衍生表型)。这种情况在建立模型时带来了几个问题,如过拟合、特征的多重共线性和系数的无限解[107]。建议在集成多组学之前进行降维。Kegerreis等[108]表明,基于wgcna共表达模块的分类[109]可以更好地应对数据集之间的差异与基于原始基因表达的分类进行比较。其他降维技术,如支持向量机(svm)、随机森林(RF)和奇异值分解(SVD)也常用于减少过拟合问题。在单细胞组学中,采用PCA、tSNE和UMAP等方法进行降维。先进的深度学习方法,如变分自编码器(VAE)也可以输出高维数据的低维潜在表示。多重测试膨胀和显著性标准是伴随问题[110]。
Heterogeneity and harmonization of the data sources
来自不同群体或联合体的样本实际上可能来自相同的受试者。例如,对来自PsychENCODE联盟中超过1800个大脑的DLPFC RNA-seq数据的综合分析[31,111]包括来自BrainSeq研究第一阶段的约500个大脑[112]。我们的大多数BRAINcode大脑样本来自Banner Health脑库,也可能在其他队列中进行研究。通过源ID(例如dbGap中的SOURCE_SUBJECT_ID)或通用ID(例如PDBP中的GUID)连接不同的队列可以通过删除重复的样本来潜在地减少偏差,并通过连接来自相同主题的样本来增加功率。当分析依赖于来自相同主题的组学时,样本识别和匹配对于某些数据集成分析至关重要,例如QTL定位。方法dram[113]提供了一种基于基因型的解决方案,以确保数据对齐。最后但并非最不重要的是,目前的单细胞组学数据在各种出版物中传播。随着越来越多的单细胞组学数据的出现,一个统一质量控制的单细胞组学数据集中存储库将有助于跨队列比较和整合。
Open data sharing
开放的基因组数据共享是成功研究的重要组成部分,这可以追溯到人类基因组计划。基因组研究产生的数据的广泛共享使数据的效用和此类项目的公共利益最大化[114]。在过去十年中,公共和私人资助机构都认识到数据共享的重要性,并敦促在数据产生之后,甚至在数据生产者首次使用之前共享数据。与大脑相关的联盟,如psychENCODE、AMP-AD、CommonMind和AMP-PD都是该政策的良好倡导者和实践者。集中的数据存储库,如Synapse (https://www.syna pse.org)和NIGADS (https://www.niagads.org)使数据共享和下载变得容易。开放共享政策也被应用于协议、方法和代码,以提高研究的可重复性[115]。
提供了一个可公开编辑的大脑多组学资源网页:http://bit.ly/brain_omics


















 202
202

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











