







一,安装所需软件
仅仅谈论蛋白直接晶体结构解析,不涉及到分子对接、分子动力学模拟等过程,需要下载的软件有CCP4、Phenix、Coot、PyMol等,蛋白质结构解析的各种软件主要在Linux系统下使用

1,CCP4套件(可包含CCP4+coot,可以在CCP4官网下载CCP4软件包,包含了Coot)
推荐是直接安装CCP4软件包,只安装Coot会缺失一些功能比如MolProbity、Clash、Ligand等,有经验的人可以自己安装这些扩展。
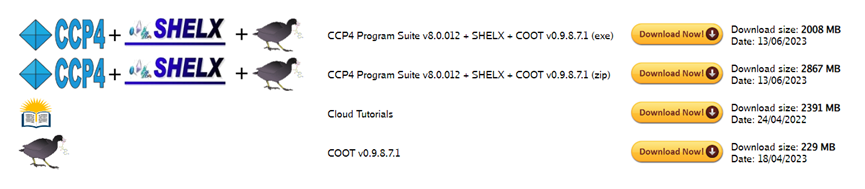
https://www.ccp4.ac.uk/download/#os=linux
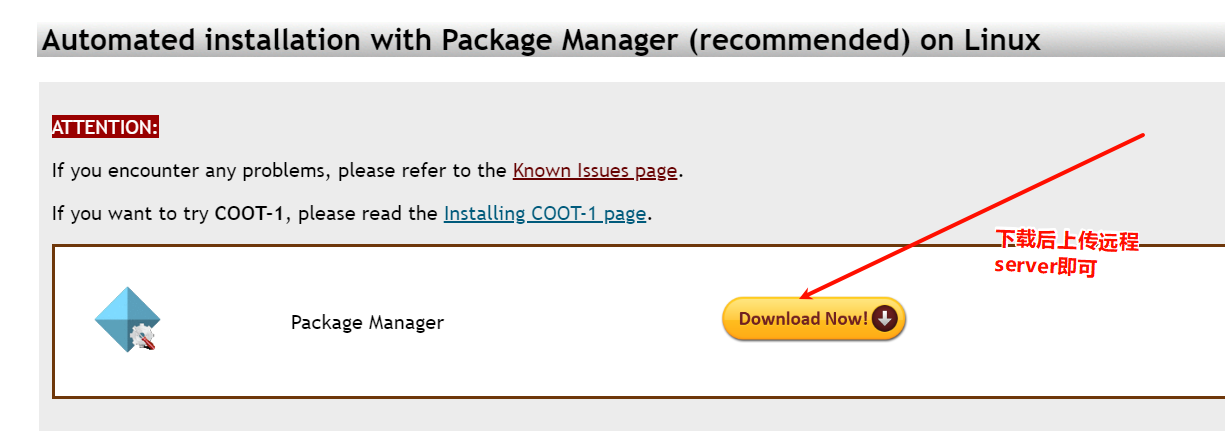
不建议在上面vscode中安装,在mobaxterm中执行,因为mobaxterm中有xserver功能
Xserver功能,可以用于在Windows上远程运行图形化Linux应用程序。
使用mobaXterm的Xserver功能,可以在Windows上打开Linux应用程序的图形界面,就像在本地计算机上一样。这使得远程工作更加方便,可以在Windows上直接运行Linux应用程序,而无需使用远程桌面连接等工具。
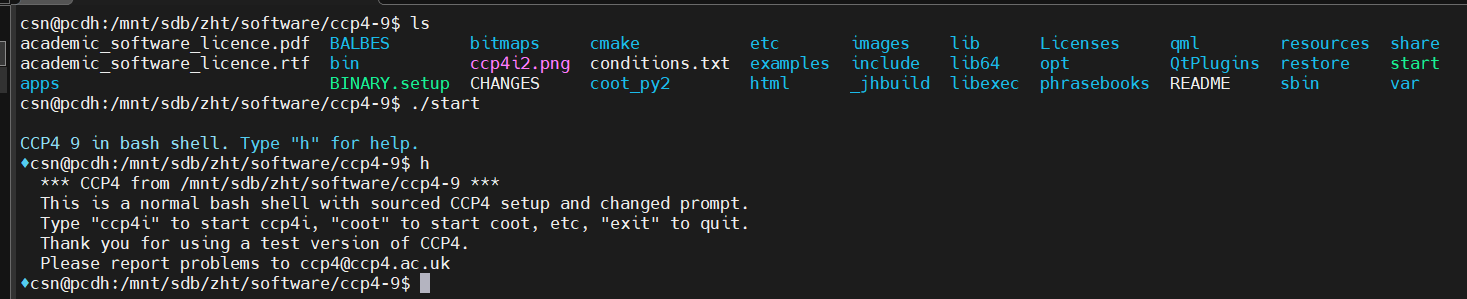
远程Linux主机是必须使用Xserver协议才能开启它的可视化界面,而后续的安装需要可视化界面一步一步操作
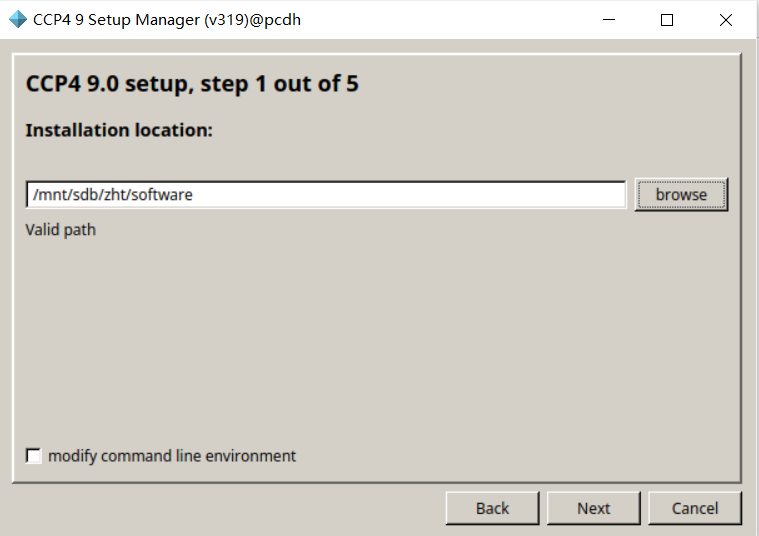
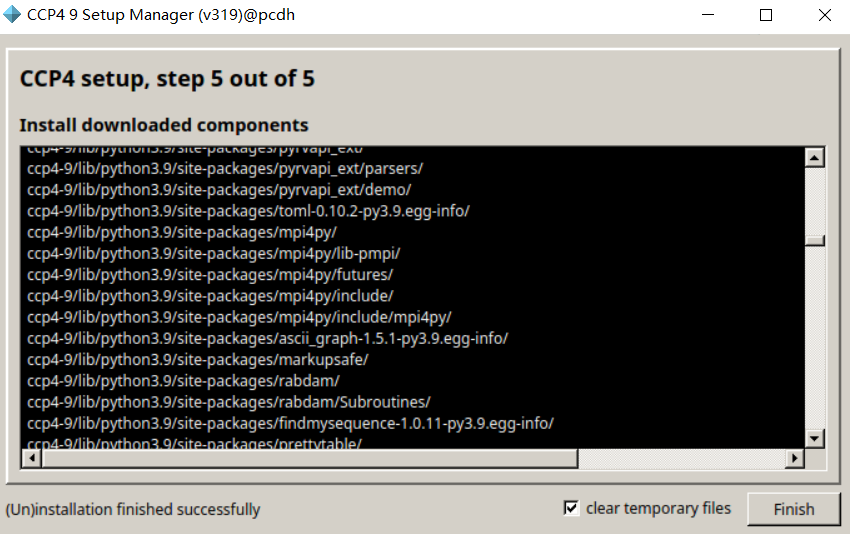
修改安装路径
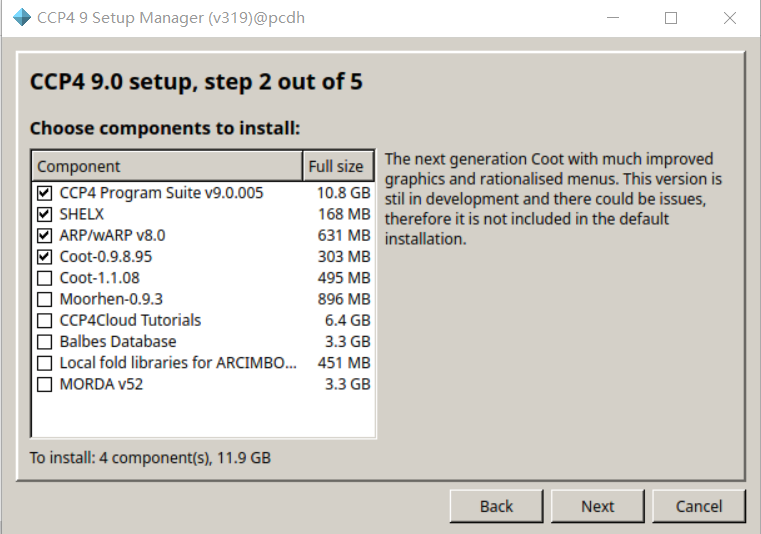
第1个CCP4核心包
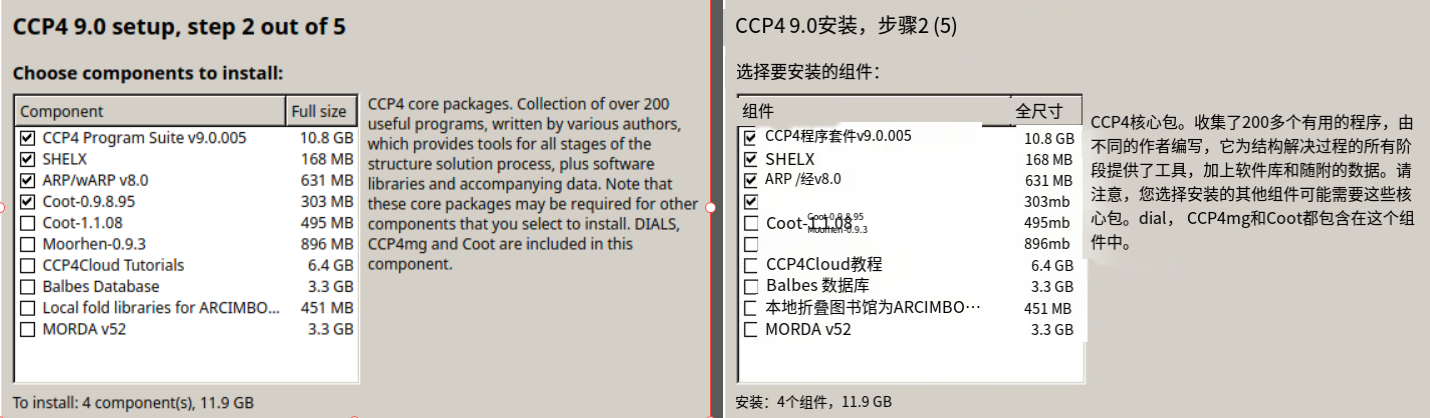
第2个SHELX套件

第3个晶体大分子构建
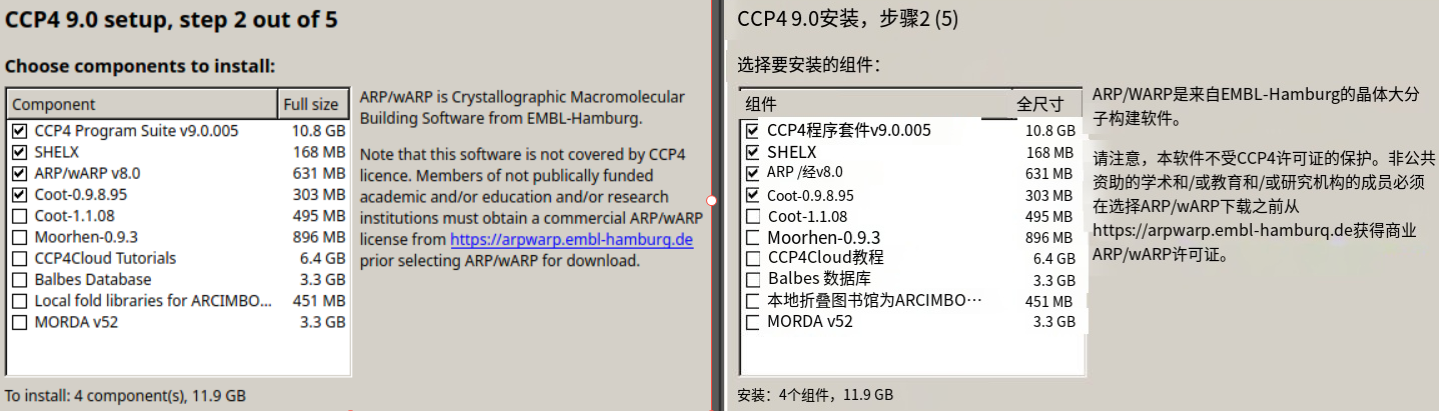
第4个就是图形交互工具coot(晶体及电镜结构可视化搭建软件)

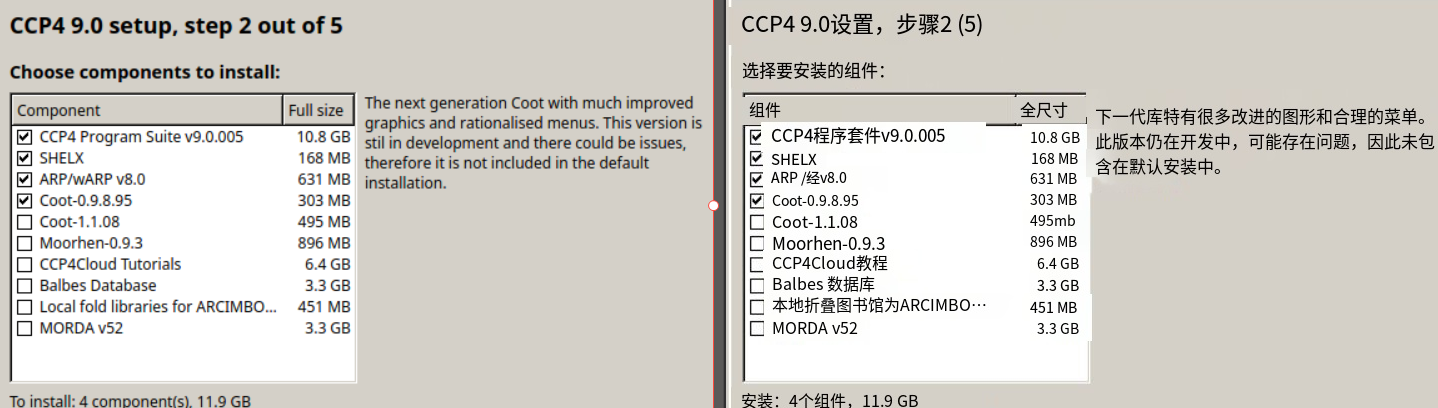
我们只安装在默认安装列表中的低版本的coot,因为高版本的仍然在开发中
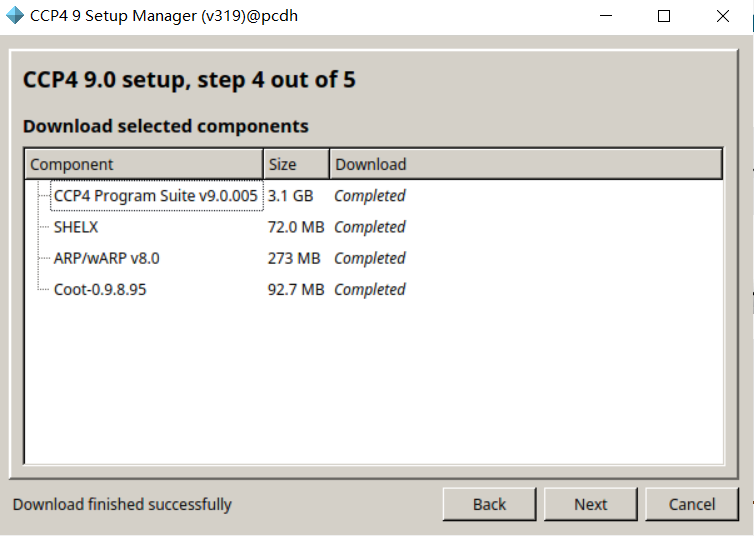
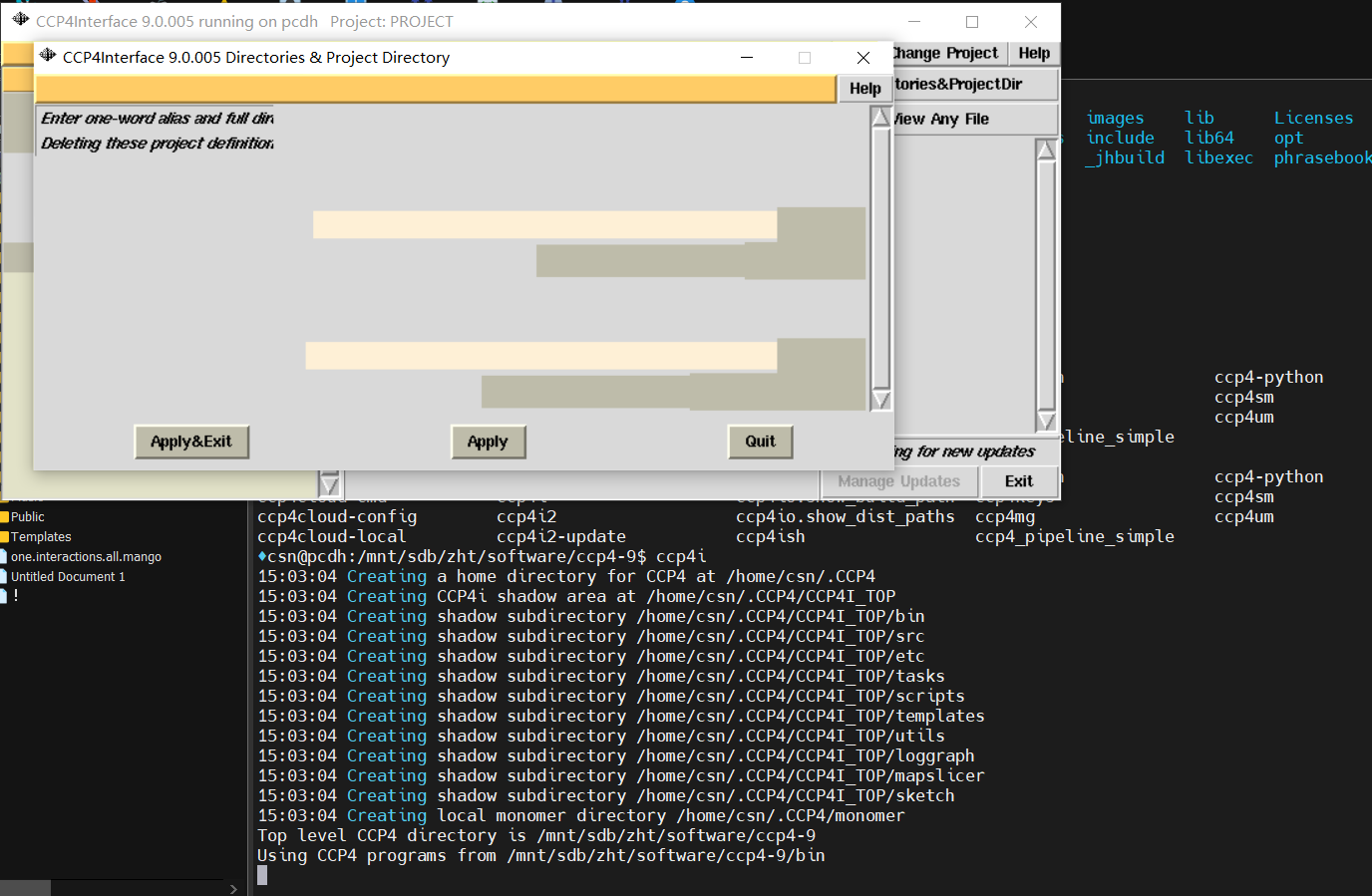
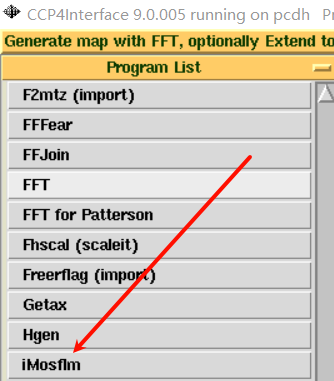
在program中可以打开iMosfilm
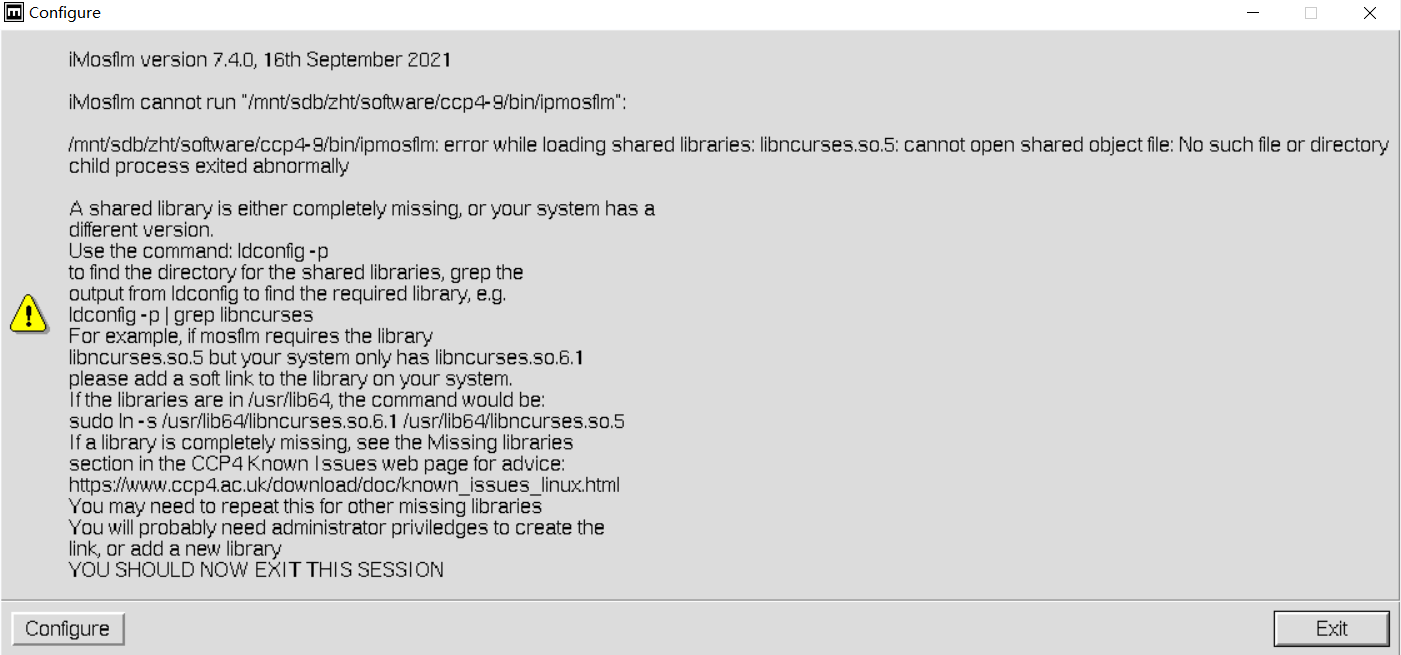
但是有问题:
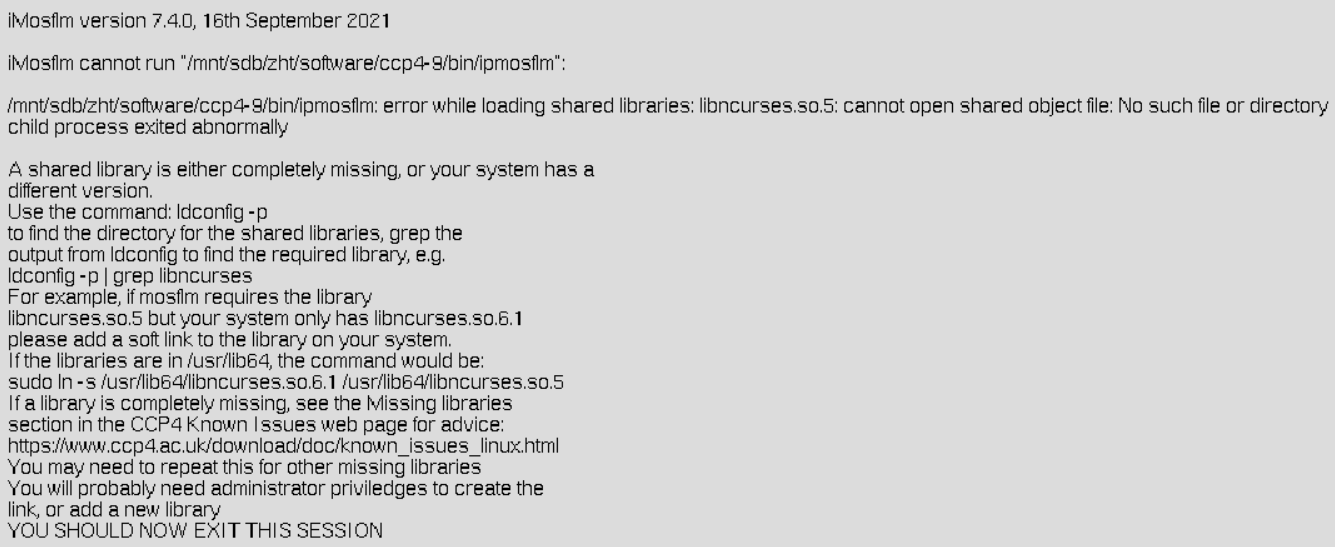
在尝试运行 iMosflm 软件时,系统无法找到所需的共享库 libncurses.so.5。这通常发生在以下几种情况:
- 库文件缺失:系统中可能没有安装该库,或者安装的版本与软件要求的版本不匹配。
- 库文件版本不匹配:系统中可能存在一个更新版本的库文件(如
libncurses.so.6.1),但软件需要的是旧版本(libncurses.so.5)。 - 库文件路径问题:库文件可能已经存在,但系统没有正确地指向它的路径。
检查库文件是否存在
使用 ldconfig -p | grep libncurses 命令来查找系统中是否存在 libncurses 库的任何版本

如果系统中存在一个更新版本的库文件,但软件需要旧版本,可以创建一个软链接(symbolic link)来指向更新版本的库文件。例如,如果 <font style="color:rgb(6, 6, 7);">libncurses.so.6.1</font> 存在,可以创建一个指向它的软链接,命名为 <font style="color:rgb(6, 6, 7);">libncurses.so.5</font>:

创建一个指向 <font style="color:rgb(6, 6, 7);">libncurses.so.6</font> 的 <font style="color:rgb(6, 6, 7);">libncurses.so.5</font> 软链接。这样,当 iMosflm 或其他程序请求 <font style="color:rgb(6, 6, 7);">libncurses.so.5</font> 时,系统会使用 <font style="color:rgb(6, 6, 7);">libncurses.so.6</font>
ldconfig -p | grep libncurses
sudo ln -s /lib/x86_64-linux-gnu/libncurses.so.6 /lib/x86_64-linux-gnu/libncurses.so.5
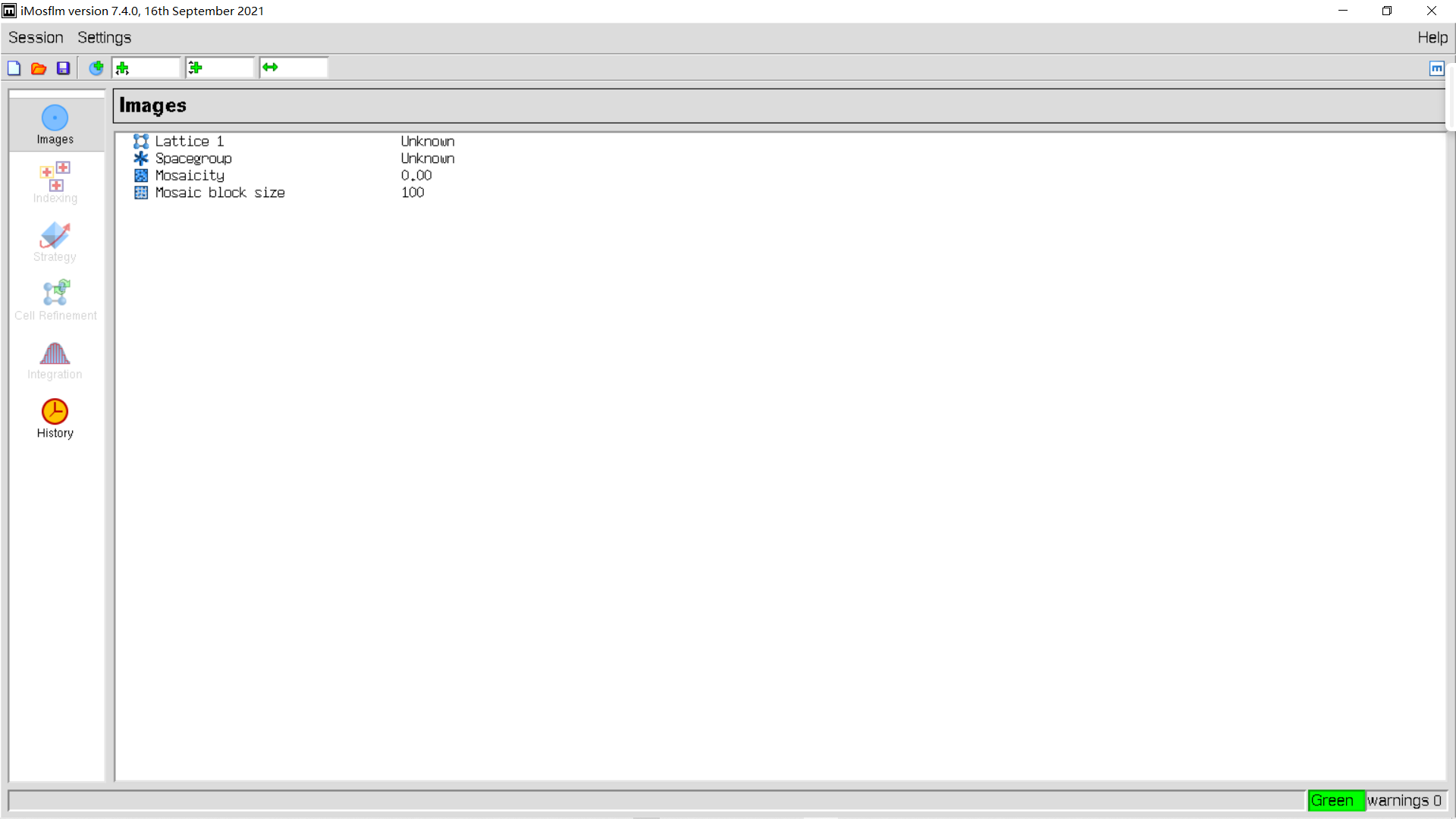
修复如下:
2,pymol安装:
https://www.pymol.org/#download
conda install -c conda-forge -c schrodinger pymol-bundle

另外需要下载一个license文件,否则一般只有30天使用期限(github上有其他平替软件,但需要自己编译)
因为基本上pymol都是图形可视化界面,所以还是需要x server,在mobaxterm中操作
3,phenix
Phenix,全称Python-based Hierarchical ENvironment for IntegratedXtallography,是一个用于晶体和冷冻电镜结构测定的综合软件包,是辅助结构搭建半自动化的软件之一

https://phenix-online.org/download

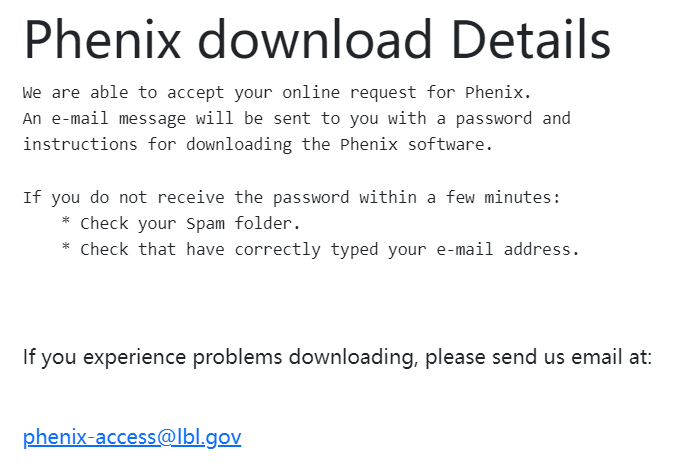
此时受到邮件:
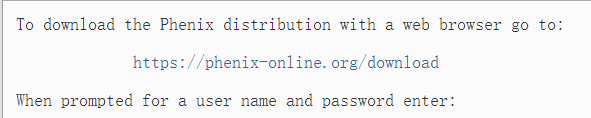
要使用网络浏览器下载 Phenix 发行版,请访问
https://phenix-online.org/download
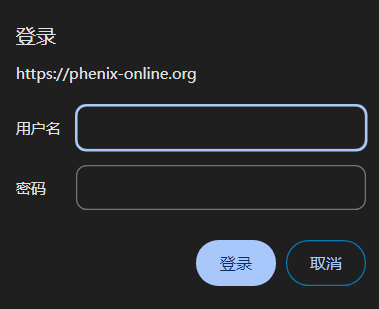
输入用户名和密码:
密码在每月 1 日 00:05 PST/PDT 时更改。
如果密码已过期,请登录 https://phenix-online.org 申请新密码。
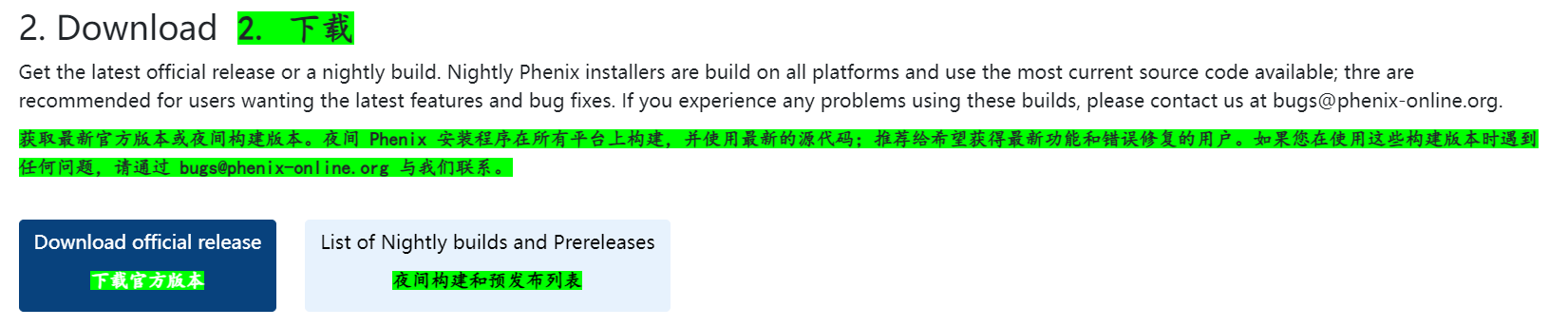
输入邮箱中获得用户以及密码,此时进入界面
https://phenix-online.org/download/phenix/release/

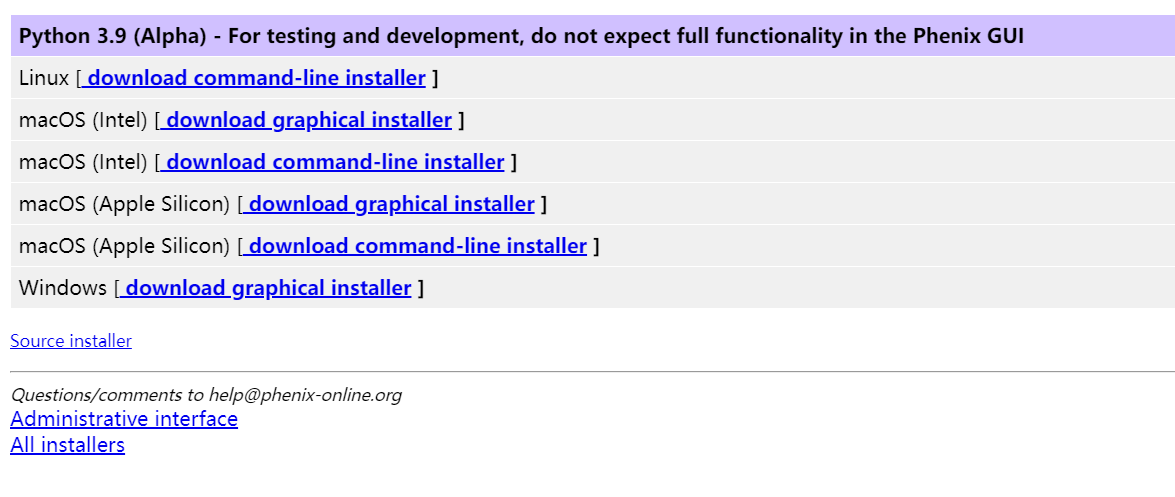
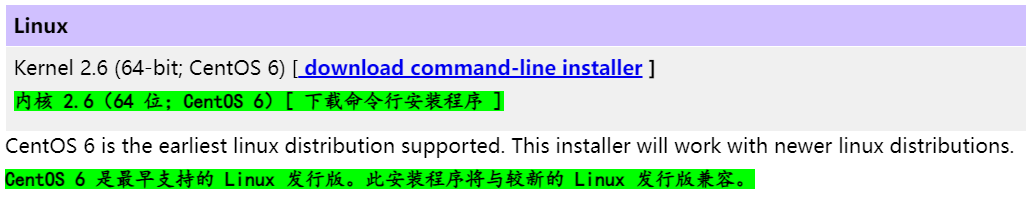
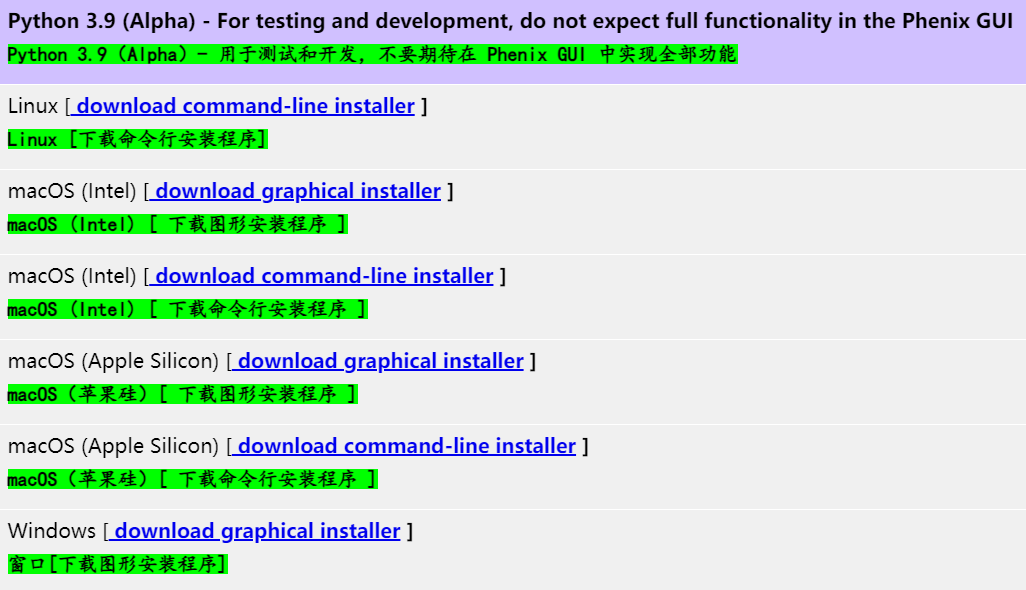
不建议使用wget或curl
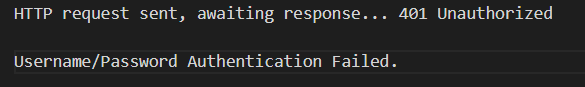
下载较慢,放在第2篇博客中介绍后续
二,蛋白质晶体结构解析所需学习内容清单:
结构生物学发展迅速并和其他学科相互渗透交叉,特别是受到结构基因组学等热点学科的极大带动。作为结构生物学的基本手段和技术,蛋白质晶体学从解析简单的蛋白质三维结构延伸到解决各类生物大分子及复合物结构,并更加注重研究结构与功能之间的相互关系,派生出诸如基于结构的药物设计等应用性很强的分支
(一)蛋白质结晶前准备
蛋白质结构功能基本介绍
提纯蛋白质,确定浓度、pH值、缓冲液等条件,控制蛋白质稳定性等。
1、目的蛋白质信息检索与调查
- 利用生物信息学工具搜集目标蛋白质的基因序列、结构域、同源蛋白质的信息
- 分析目标蛋白质的理化性质,如分子量、等电点、聚合程度、稳定性等
2、质粒制备
- 设计引物,克隆目标基因到表达载体
- 转化表达宿主,提取重组质粒
- 质粒测序验证目标基因插入
3、蛋白质纯化
- 选择合适的诱导条件,表达可溶性或不溶性重组蛋白
- 裂解菌体,释放重组蛋白质
- 蛋白质纯化:亲和层析、离子交换层析、凝胶过滤等
4、蛋白质不表达和包涵体问题
- 分析不表达的原因,优化诱导条件
- 改进溶解缓冲液条件,提高蛋白从包涵体中释放
5、蛋白质活性鉴定
- 进行Western Blot或酶活性实验验证蛋白质活性
6、蛋白质结晶前分析
- 测定蛋白质的纯度、聚合状态、稳定性
- 优化缓冲液条件,调整蛋白质到适宜的pH和离子浓度
(二)蛋白质结晶与衍射数据收集
利用协同结晶筛选获得蛋白质结晶,在同步辐射光源下收集衍射数据。
1、蛋白质结晶
- 蛋白质结晶的基本原理
- 蛋白质结晶的影响因素
- 蛋白质结晶的基本方法
- 结晶条件筛选策略
2、SSRF(同步辐射光源) 的介绍
- SSRF简介
- SSRF的光源优势
- SSRF的实验站介绍
3、蛋白质晶体衍射数据收集
- X射线结晶学基本原理
- 晶体探针和晶体定位
- 晶体测试和优化
- 衍射数据收集参数设定
- 衍射数据处理和分析
(三)蛋白质晶体结构解析软件安装
安装相关计算机程序,如Phenix, XDS, Pymol等用于后续的数据处理与模型建立。
1、Linux系统安装
- Linux系统选择和安装
- Linux系统基本命令
- Linux系统环境配置
2、蛋白质晶体结构解析软件安装
- CCP4安装
- Phenix安装
- Coot安装
- PyMol安装
- 其他结构解析支持软件安装
蛋白质结构解析的各种软件主要在Linux系统下使用。建议使用Linux系统。
首先需要对Linux系统进行简单的介绍,包括选择发行版本、基本命令使用、环境变量配置等。然后依次介绍CCP4、Phenix、Coot、PyMol等主要的结构解析软件的下载、编译和安装方法。也可以介绍一些结构解析中需要的其他软件工具的安装。
掌握在Linux系统上配置蛋白质结构解析的软件环境。
(四)Index、integrate与scale软件使用和介绍
利用软件index及integrate衍射点,scale衍射数据以校正强度。
1、晶体结构学知识
- 晶体学中的衍射理论基础
- 布拉格定律和倒易格向量
- 晶体的对称性
2、蛋白质晶体结构解析流程
- 蛋白质的表达与纯化
- 蛋白质的结晶
- X射线晶体学数据收集
- 晶体结构解析流程概述
3、Index和integrate
- Indexing的目的和原理
- Integration的目的和过程
4、Scale
- Scale的目的——校正数据
- Scale常用方法
5、使用XSCALE功能进行scale
- XSCALE软件介绍
- 使用XSCALE进行数据scale的步骤
6、使用HKL2000进行index、integrate和scale
- HKL2000软件介绍
- 使用HKL2000进行indexing
- 使用HKL2000进行integration
- 使用HKL2000进行scaling
(五)分子置换、构建优化与结构提交
利用分子置换法确定蛋白质框架,手动构建余下结构,进行优化后提交蛋白质坐标库。
1、分子置换
(1) 分子置换的概念
(2) 分子置换的目的
(3) 常用的分子置换软件介绍
(4) 分子置换的具体操作步骤
2、蛋白质晶体结构构建
(1) 蛋白质序列比对确定构建起始模型
(2) 主链构建方法
(3) 侧链构建方法
(4) 构建完成后的模型检查
3、蛋白质晶体结构优化
(1) 能量最小化原理
(2) 模拟退火原理
(3) 分子动力学模拟原理
(4) 优化过程中的评估标准
4、蛋白质晶体结构验证
(1) Ramachandran图分析
(2) 各类键长和键角分布
(3) 密接点分析
(4) B因子分布
(5) 电子密度匹配度评价
5、蛋白质晶体结构提交到PDB
(1) PDB数据提交要求
(2) 各项验证确认无误后压缩需提交文件
(3) 在PDB网站提交表单,上传文件,等待审核结果
(六)蛋白质晶体结构展示
利用Pymol等软件分析并展示蛋白质的二级结构、三级结构,活性口袋等结构信息。
1、pdb格式文件简介
- pdb文件概述:包含蛋白质晶体学数据的标准格式
- 原子坐标:记录每个原子的xyz坐标
- 温度因子:记录每个原子的热运动参数
- 二级结构:记录α螺旋和β片层的位置
- 结构注解:记录配体、酶活性中心等重要结构信息
2、PyMOL制作蛋白质晶体结构图
- PyMOL简介:流行的分子可视化软件
- 加载pdb文件
- 显示蛋白质链、α螺旋和β片层
- 调整视角、变色和放大关键结构
- 导出高质量图像
3、使用PyMOL制作蛋白质配体结合位点信息
- 识别蛋白质与配体的相互作用
- 突出显示配体结合位点残基
- 在结合位点生成表面模型
- 制作配体结合位点的特写图
4、使用PyMOL调查蛋白质的温度因子B-factors
- 显示温度因子putty图
- 分析柔性域和稳定域
- 与酶活性中心和功能位点的关系
5、使用PyMOL重叠对比不同的蛋白质晶体结构
- 载入不同状态的pdb文件
- 重叠对齐蛋白质结构
- 比较构象变化,如酶动力学过程中的不同中间状态
6、使用PyMOL显示蛋白质晶体结构中配体的电子密度图
- 加载包含配体密度的pdb文件
- 显示2Fo-Fc 和 Fo-Fc电子密度图
- 检查配体与电子密度的匹配程度
- 评估配体定位和取向的准确性
7、使用PyMOL结合Chimera实现同步显示非对称单元的蛋白质分子
- 在PyMOL中显示蛋白质非对称单元
- 在Chimera中同步显示非对称单元
- 细节对比不同分子中的相同结构
- 分析蛋白质多聚体形成的分子间相互作用
参考:

















 1769
1769

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









