







参考我的上一篇博客:
https://blog.csdn.net/weixin_62528784/article/details/144830955

文件如下:

一,phenix方面:
1,新建文件夹处理:
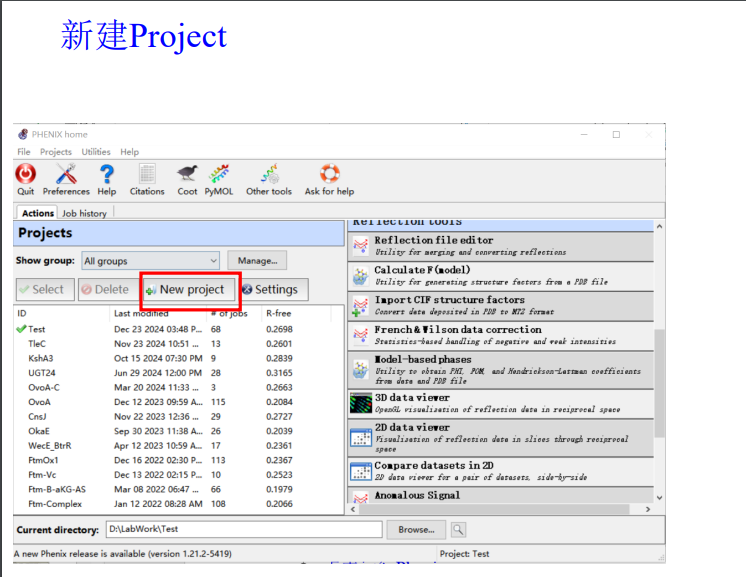
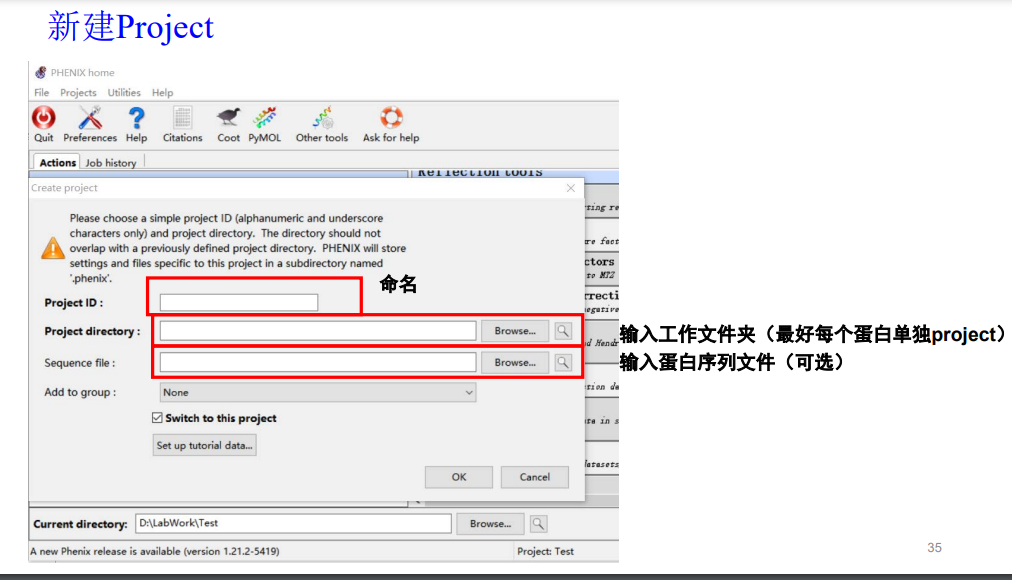
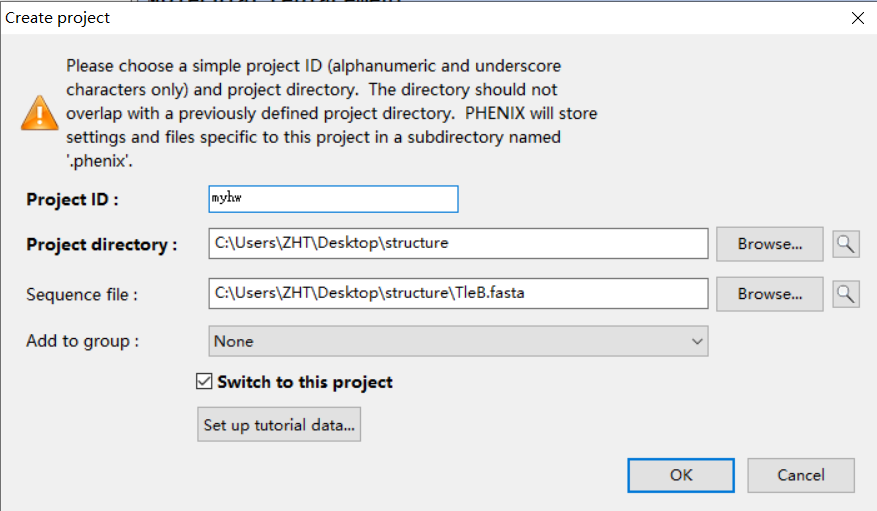
项目名称:hw(homework)
工作文件夹:此处放置在含有mtz文件的文件夹内
序列文件也选上
总体上而言:我们精修的是TleB蛋白质,目前已经有了X射线晶体衍射结构以及相关pdb模型
也就是
2,运行Xtriage程序
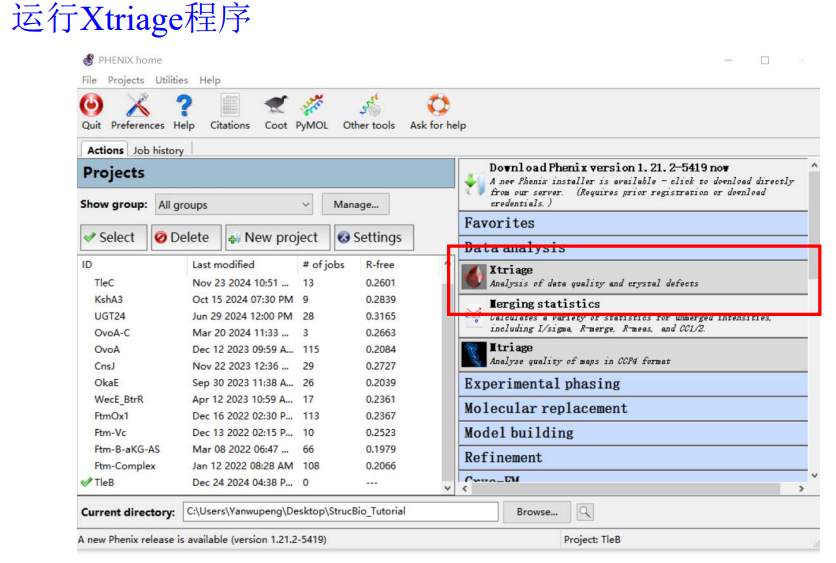
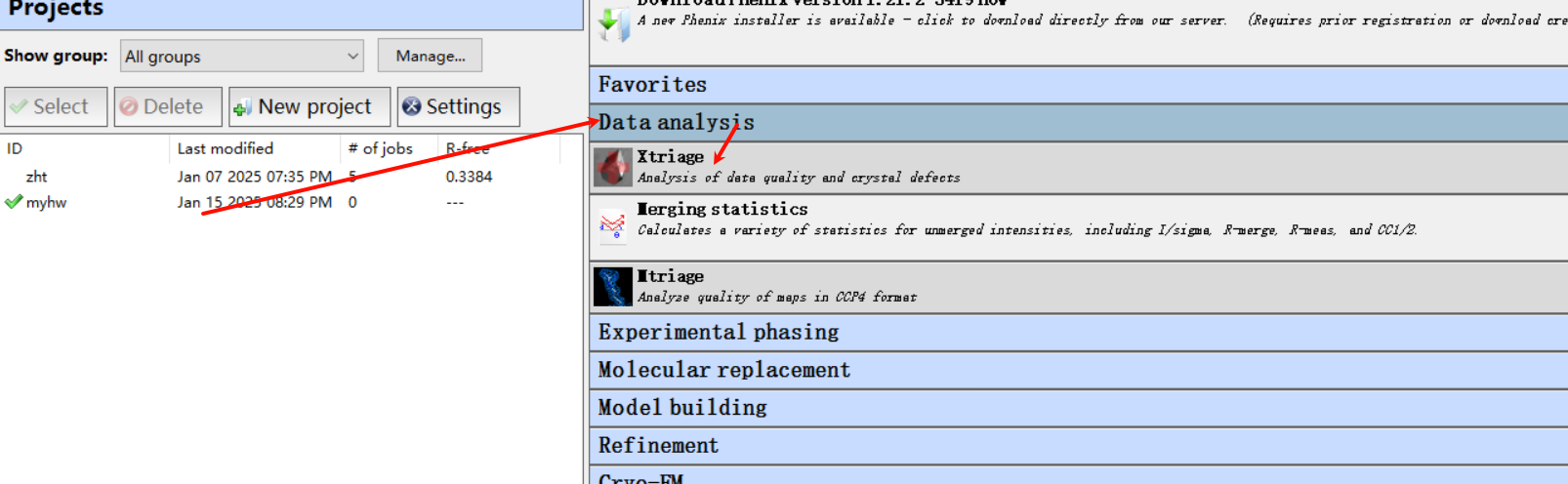
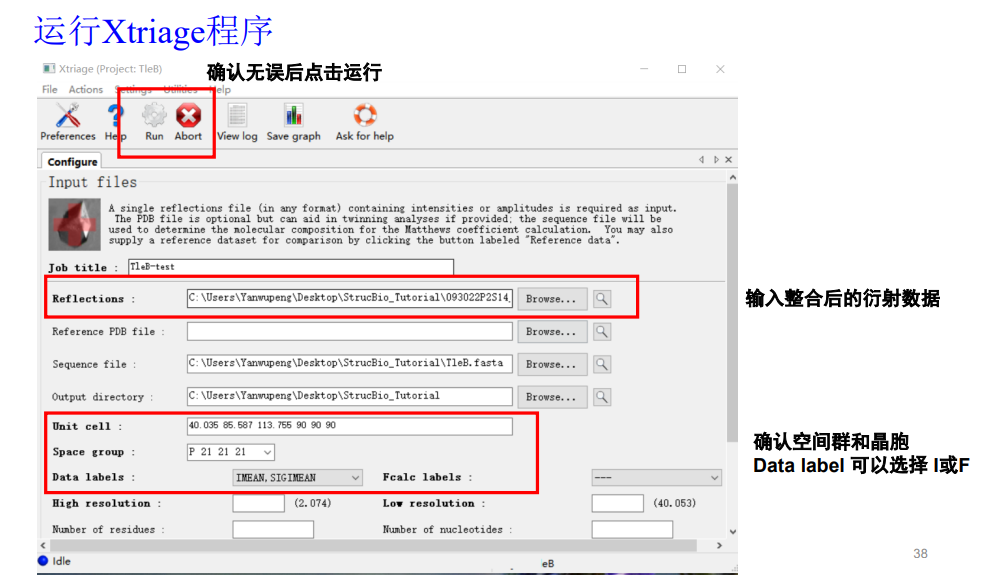
大概配置如下:
第一行的衍射数据就使用前面那个文件夹里的mtz文件,序列文件也一并用上(就是TleB该蛋白的)
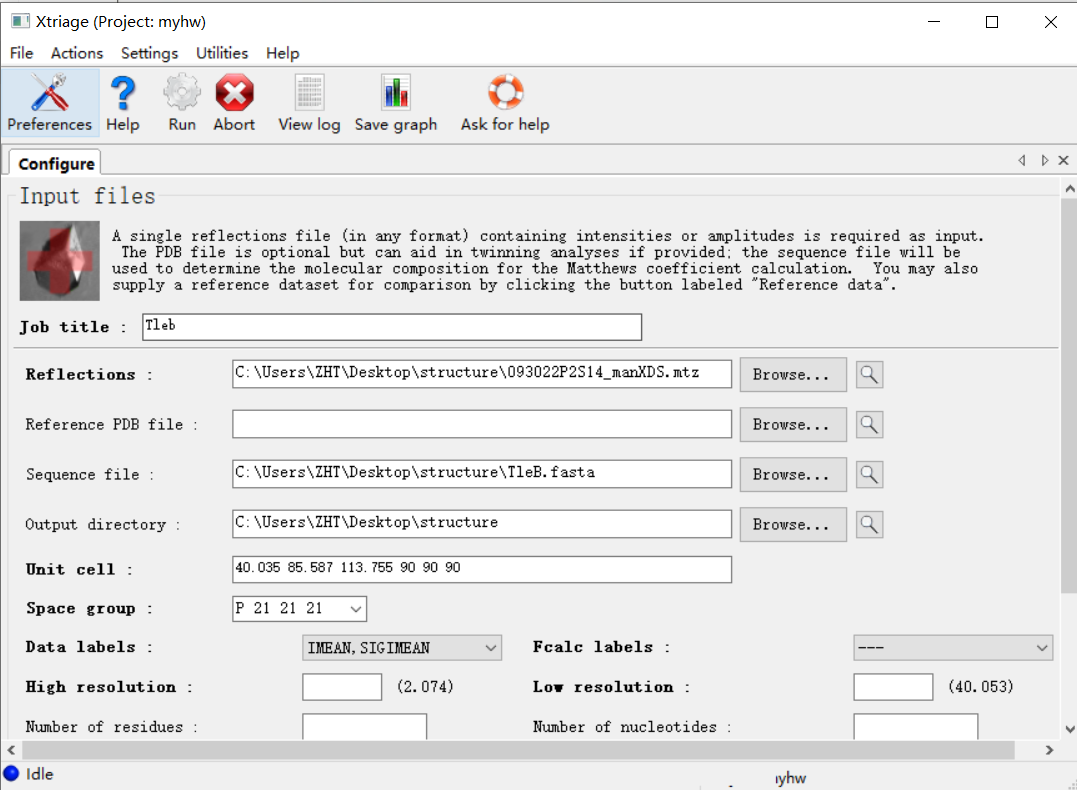
点击确认之后就在运行:
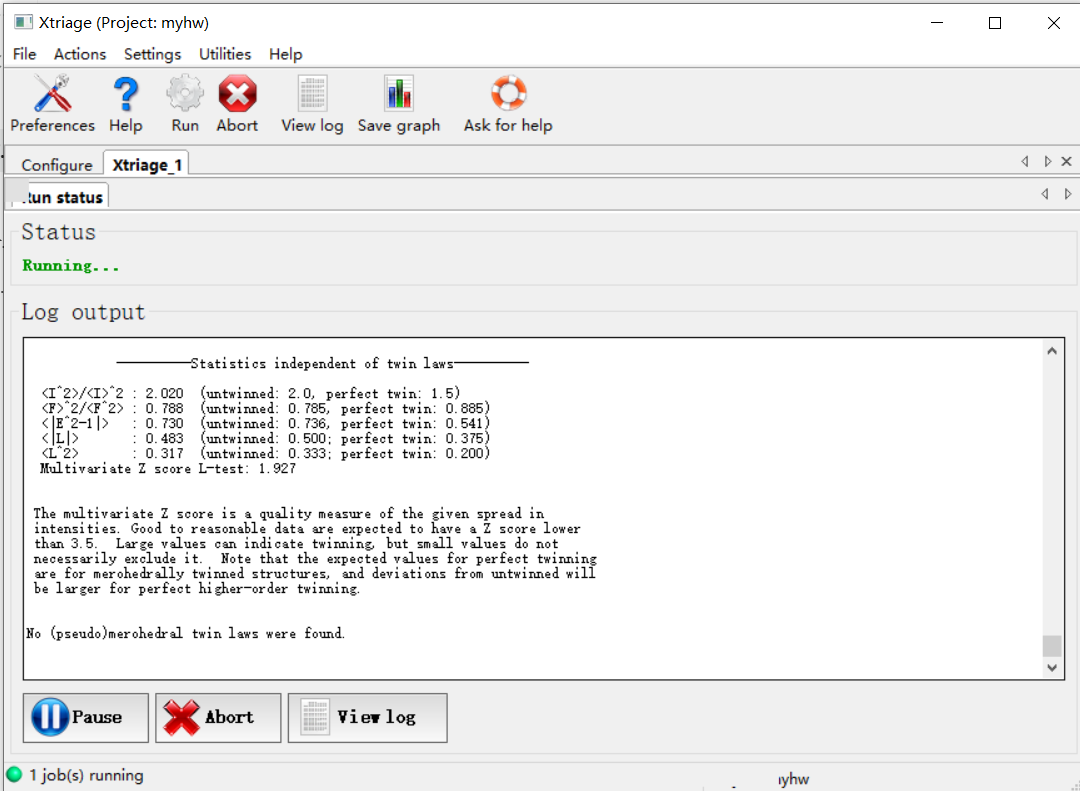
对于输出结果,有很多指标可以查看:
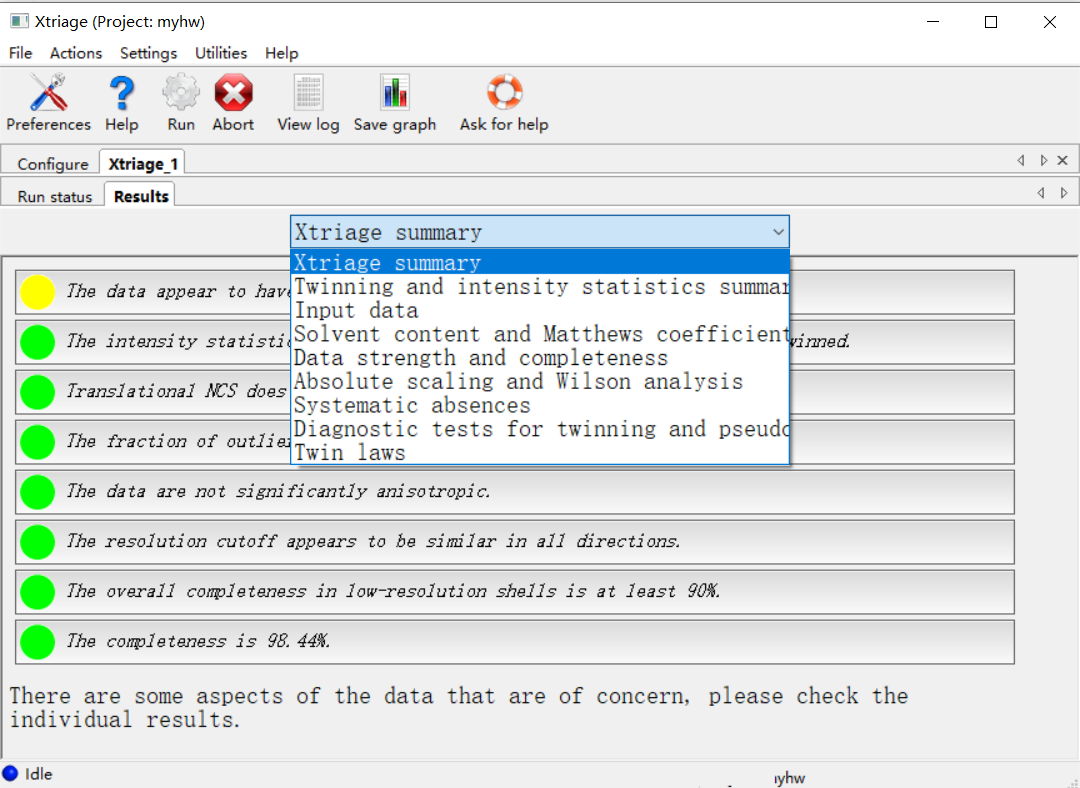
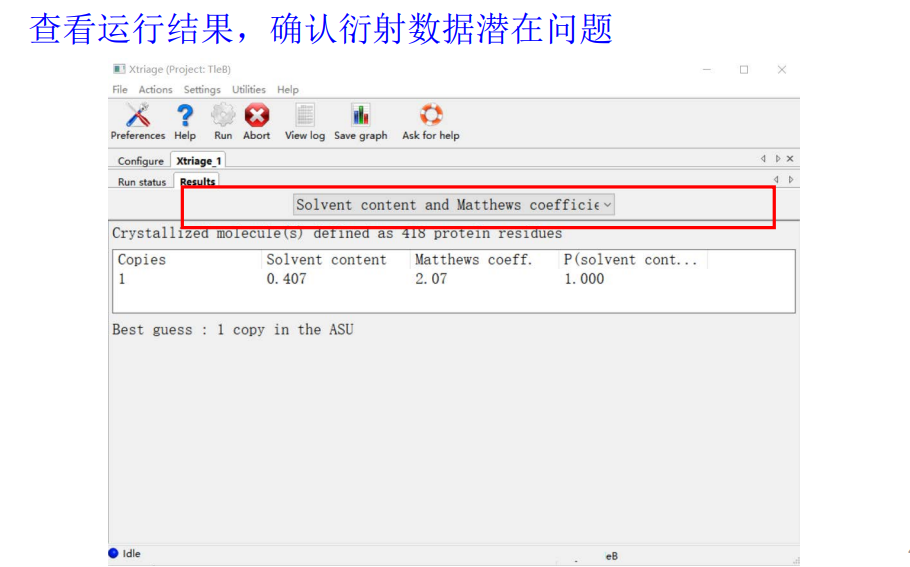
有很多其他的条目可以翻阅: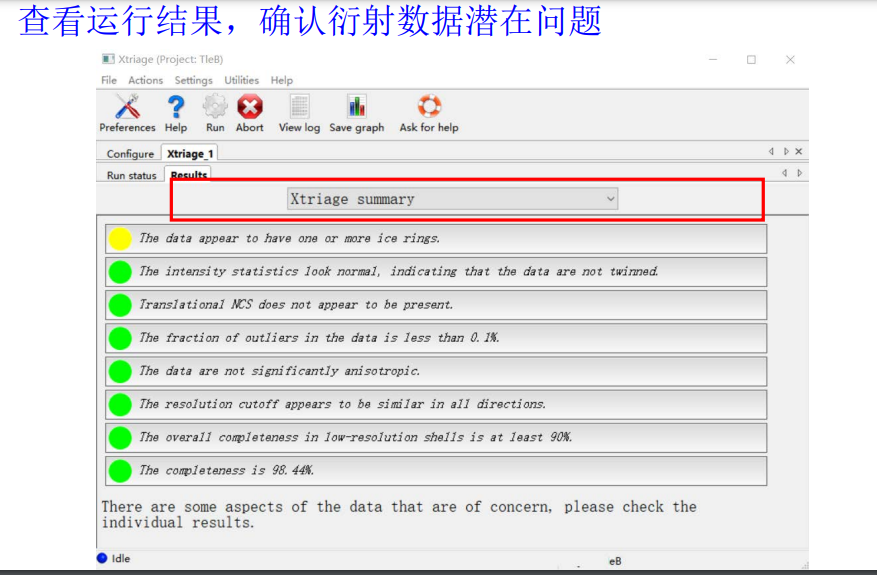
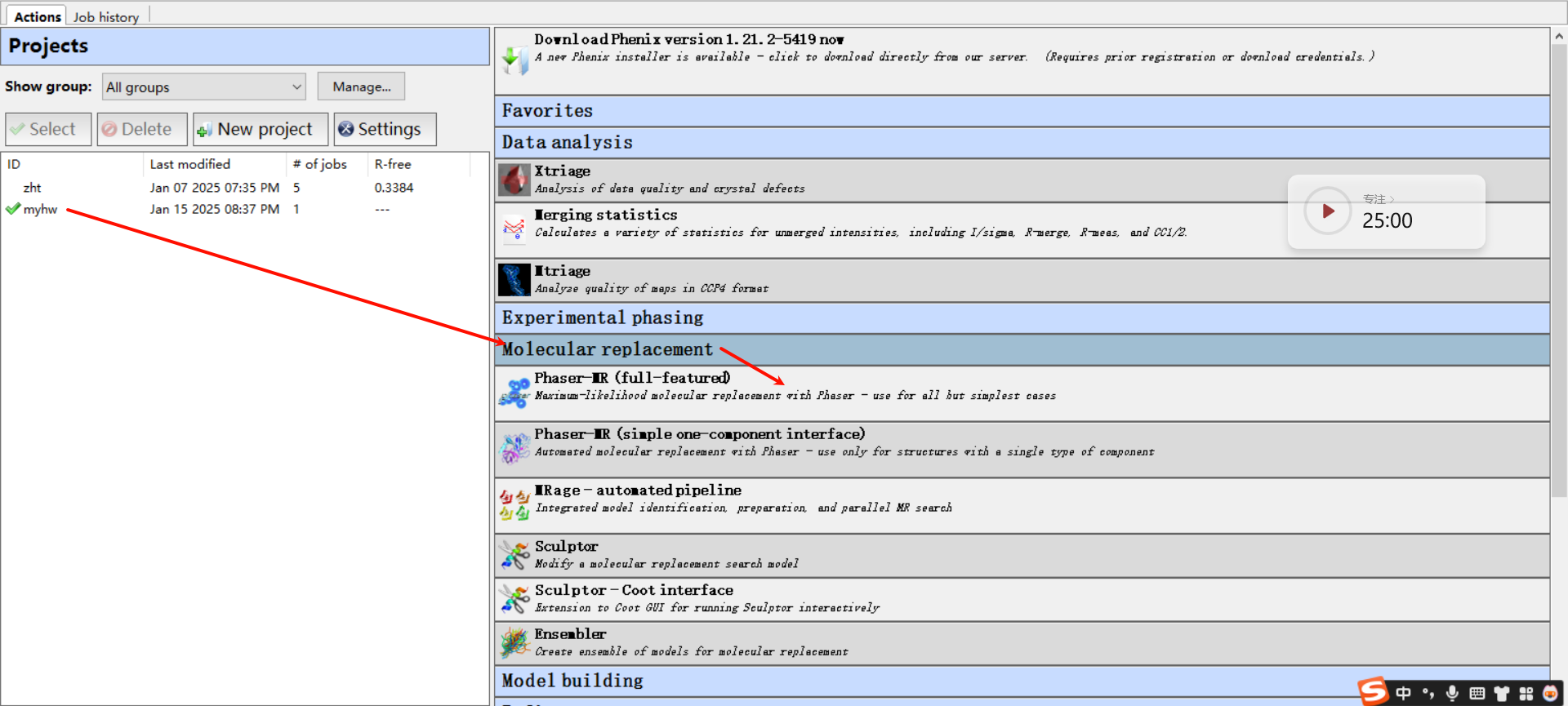
3,运行Phaser程序:回到phenix home界面
注意是分子置换的选项界面内
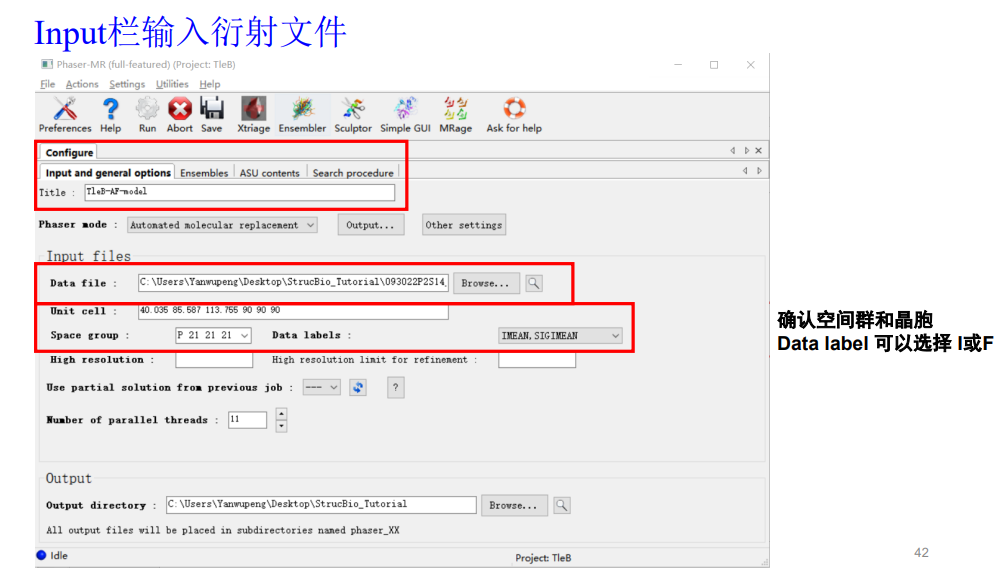
同样的衍射数据选择mtz那个文件,和前面一样
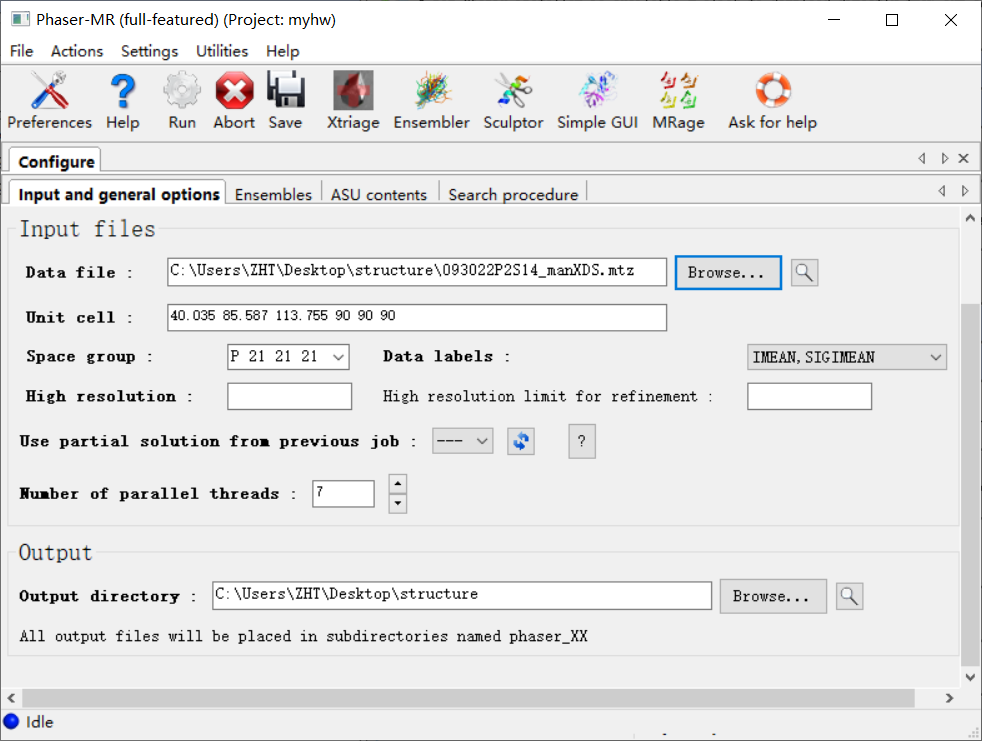
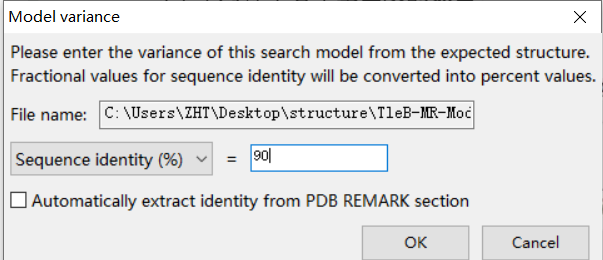
接着进入第2个选项ensembles:
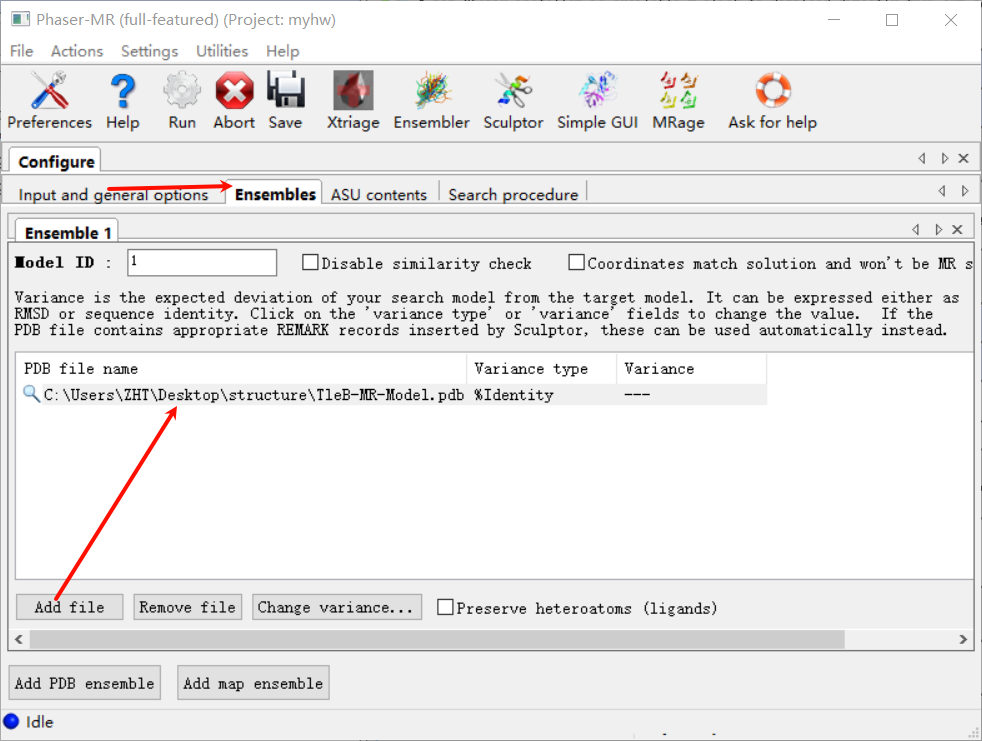
选择文件夹中的那个pdb文件
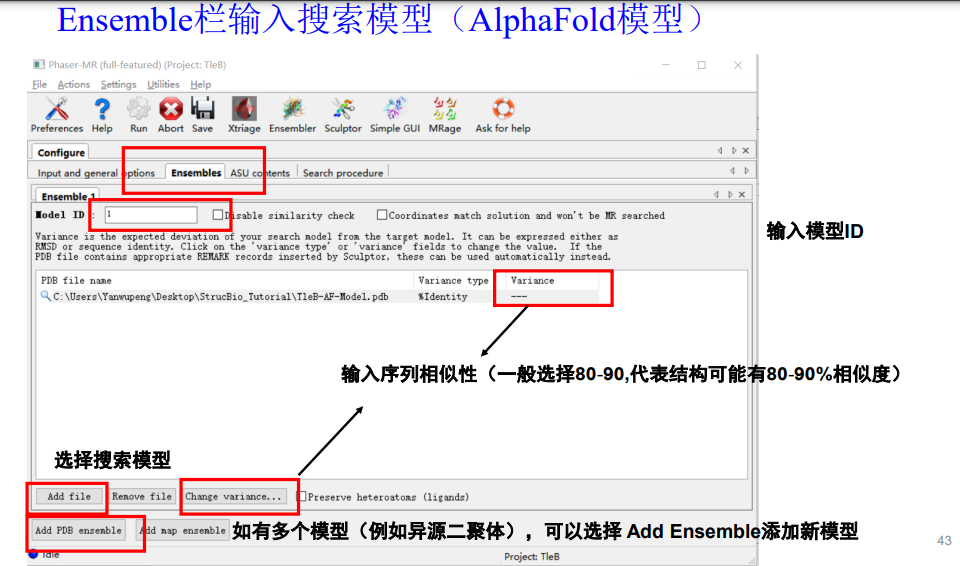
注意!这里的序列相似性不要选择100%,即使是同个蛋白用AF3预测的结构,也要给一点自由度
整体如下:
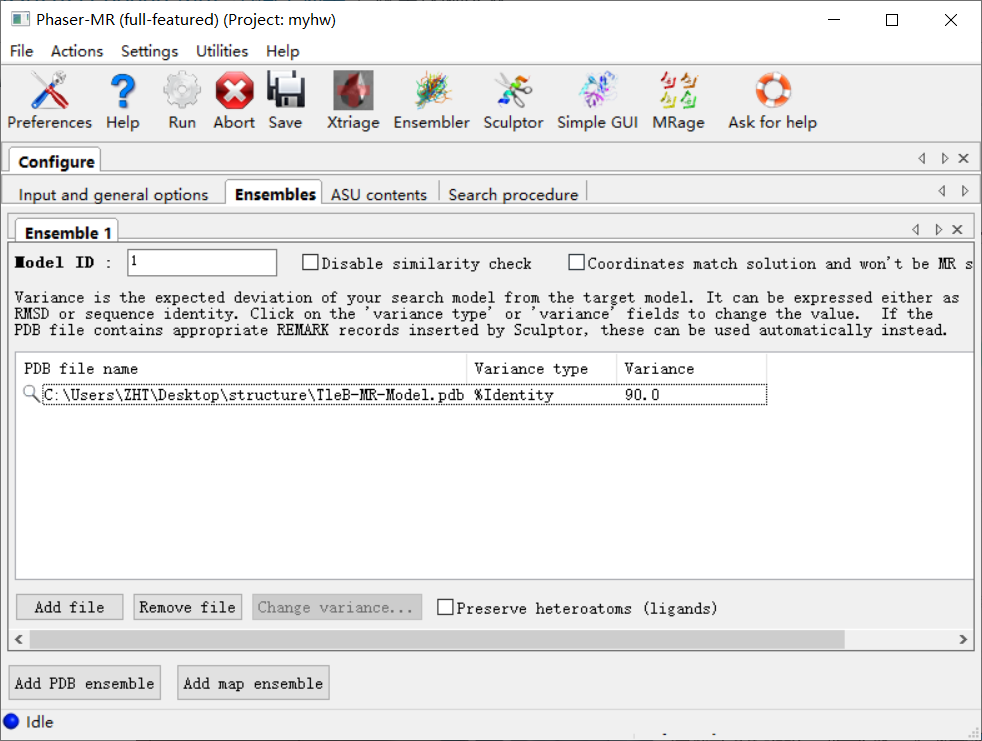
然后接着是第3栏:
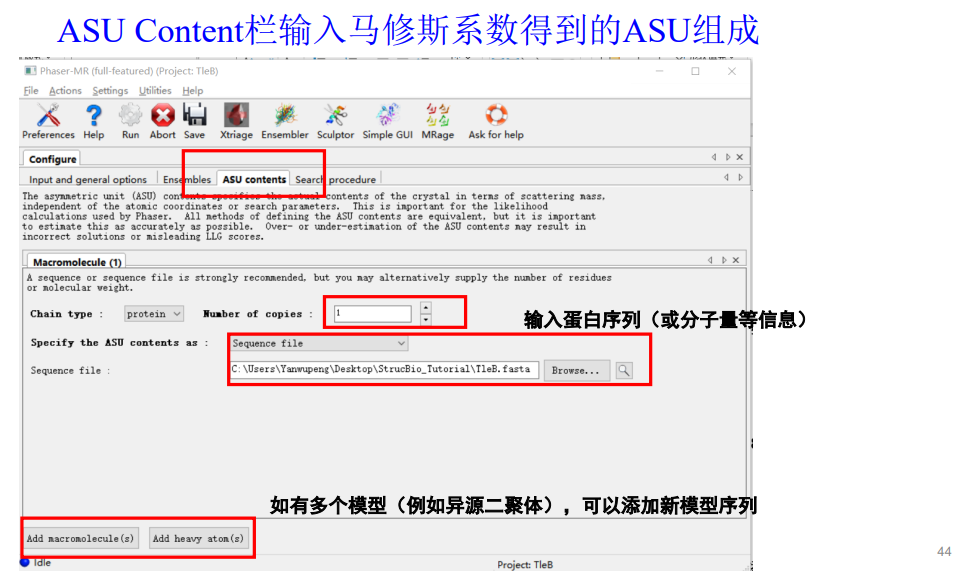
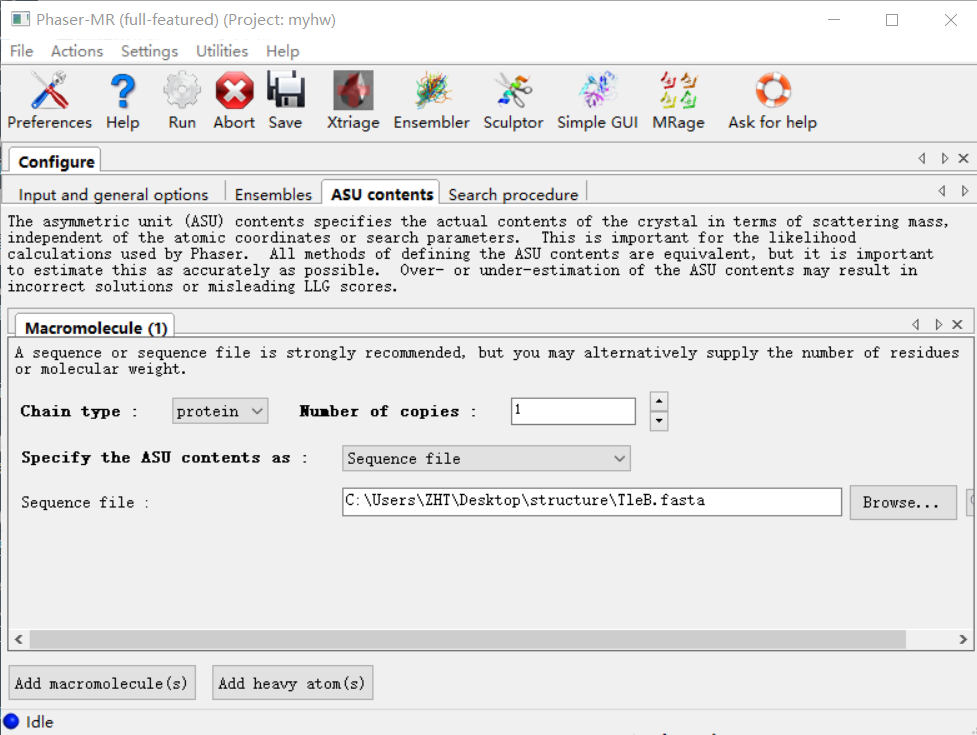
再接着是第4栏:
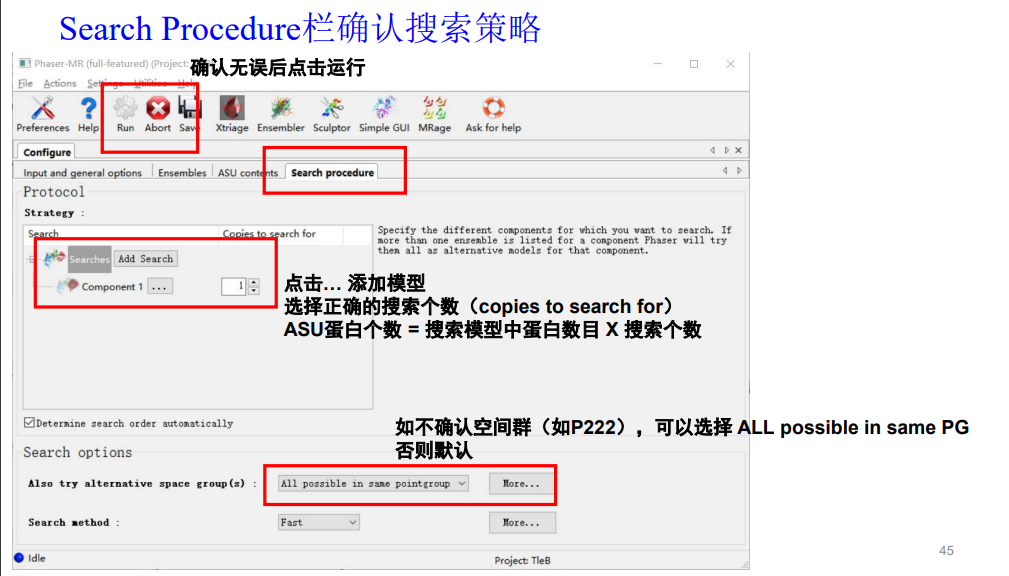
操作如下:
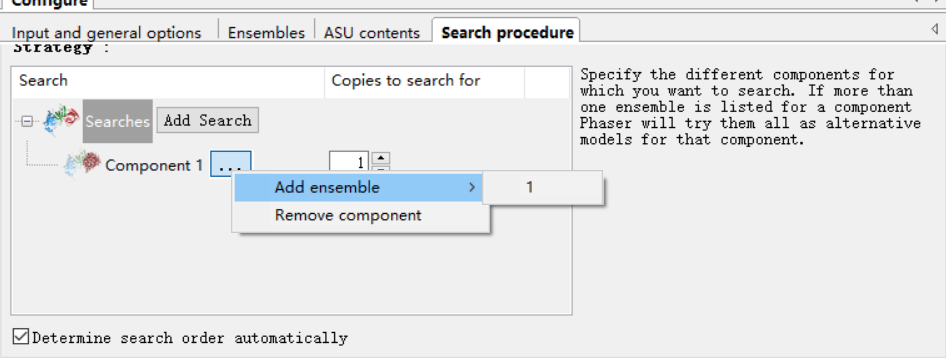
我的模型中蛋白的数目x搜索的copy=每个不对称中心中蛋白质的数目
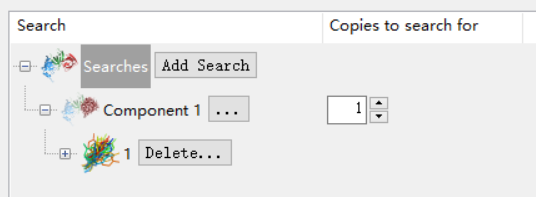
然后我十分确认就是这个空间群,所以不用浪费时间了,我就直接选择No alternatives
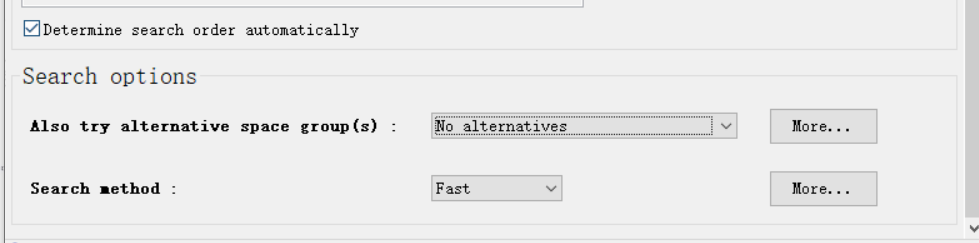
然后就是直接run
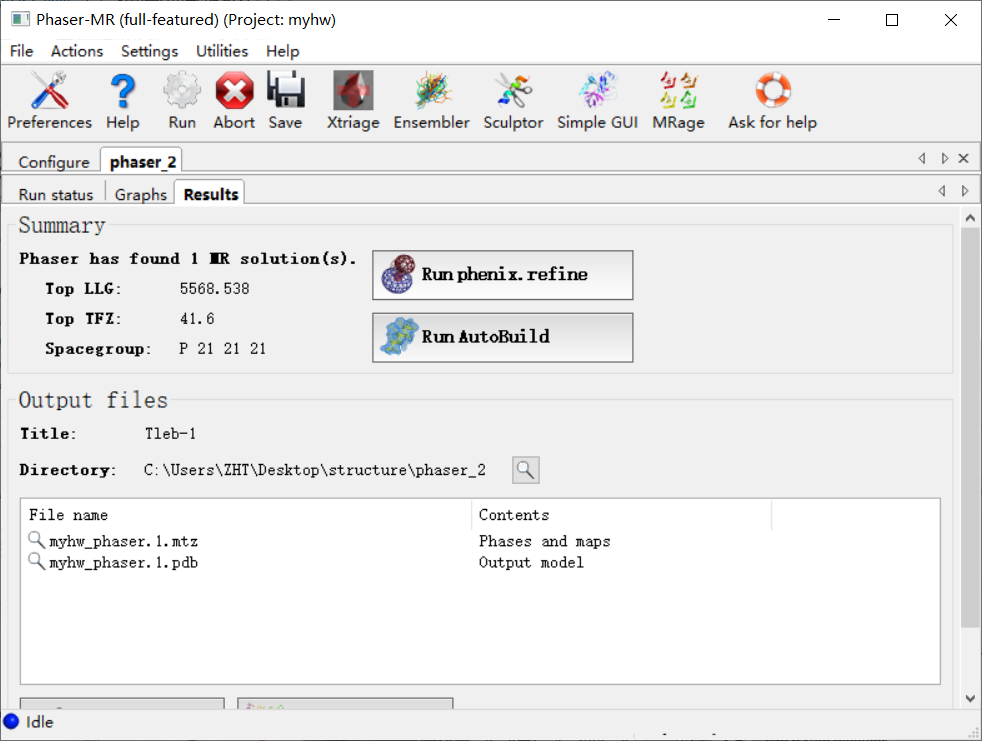
然后运行结果如下:
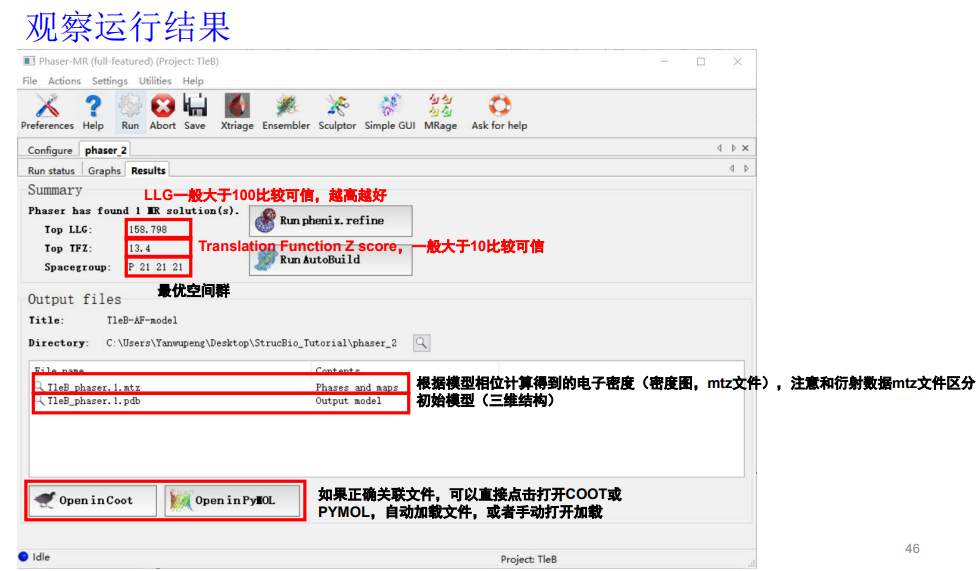
TFZ最好大于10,分子置换的结果就八九不离十了,说明后续精修不会太费力了
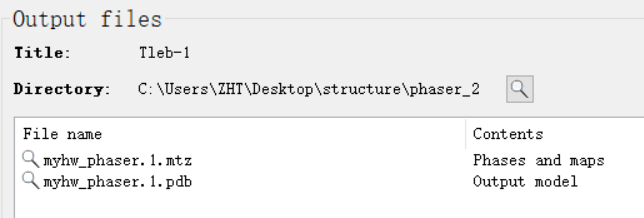
注意输出的位置以及文件:
4,在分子置换之后可以使用coot进行精修,同时查看结果
没有关联好wincoot的路劲,就需要自己导入这个模型结果了(mtz以及pdb)

注意在coot中是打开分子置换之后的pdb以及mtz文件
pdb是结构文件,mtz是经过分子置换之后得到的分子密度文件
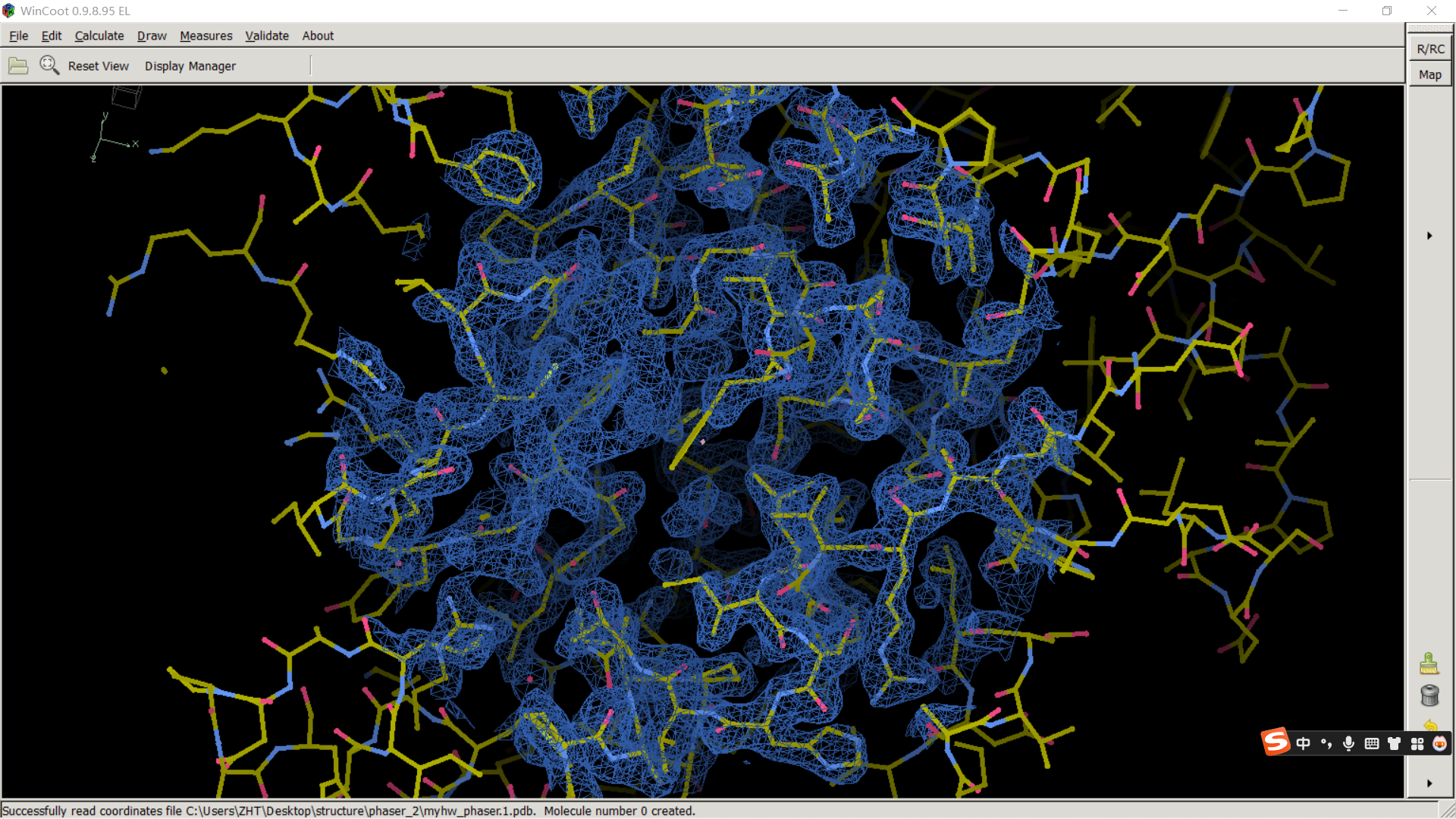

蓝色的电子密度其实就是2Fo-Fc,
如果是绿色的电子密度就是Fo-Fc(Fo是observation,观测、实验、衍射数据;Fc是calculation,根据模型计算出来的)
如果Fo-Fc>0,代表着Fo是有东西的,Fc是没有东西的,也就是说明我们的模型里没有东西,但是实际观测是能看到东西的,也就是绿色的;
红色的正好相反,就是Fo-Fc<0,观测obs是看不到东西的,但是我们的模型在这里有东西;
所以我们的模型精修理论上就是将红色的以及绿色的电子密度都搞没,然后理想情况下都是蓝色的;
其实可以看到,我们上面的模型就是几乎全部都是蓝色的
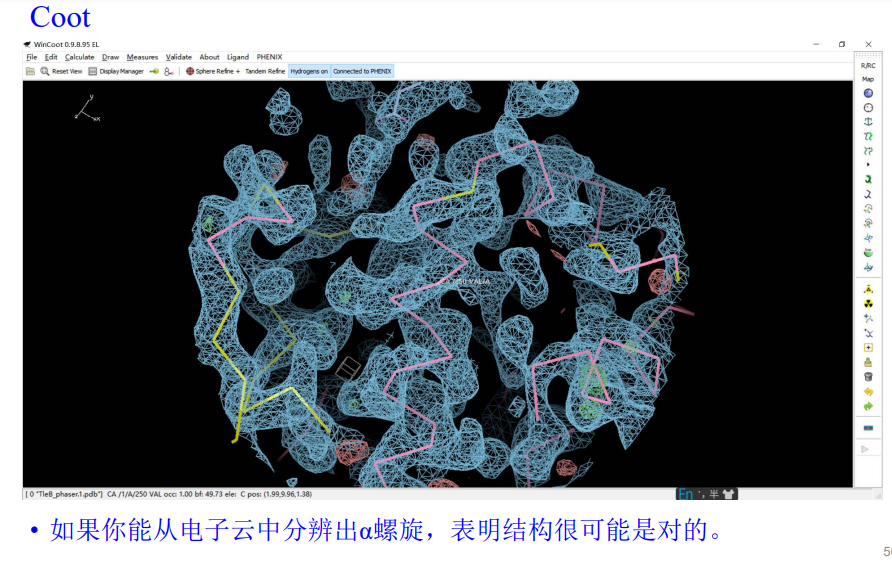
5,在获得分子置换的结果之后,首先不是人工精修,一般是先机器精修一次
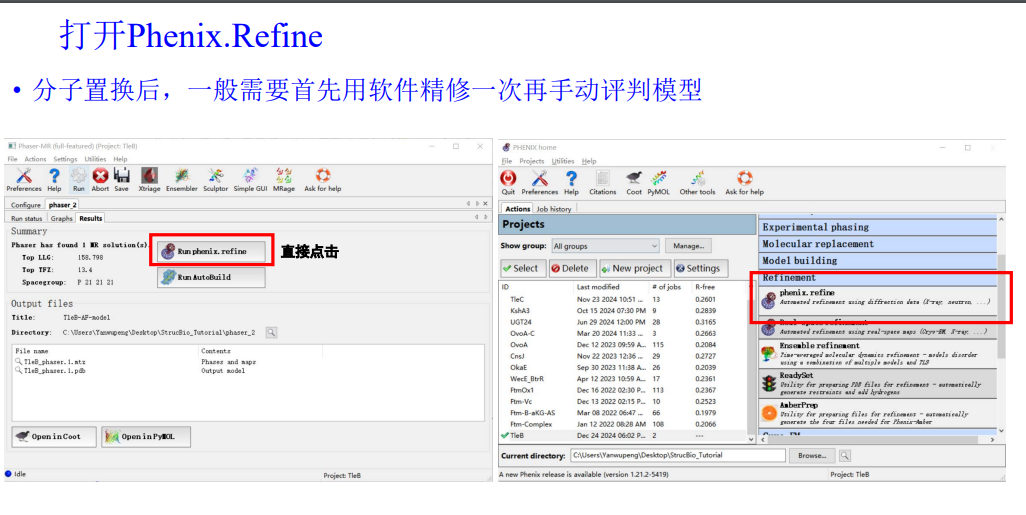



实操:机器精修开始
其实就是原来分子置换结果右上角对应的refine的图标位置处
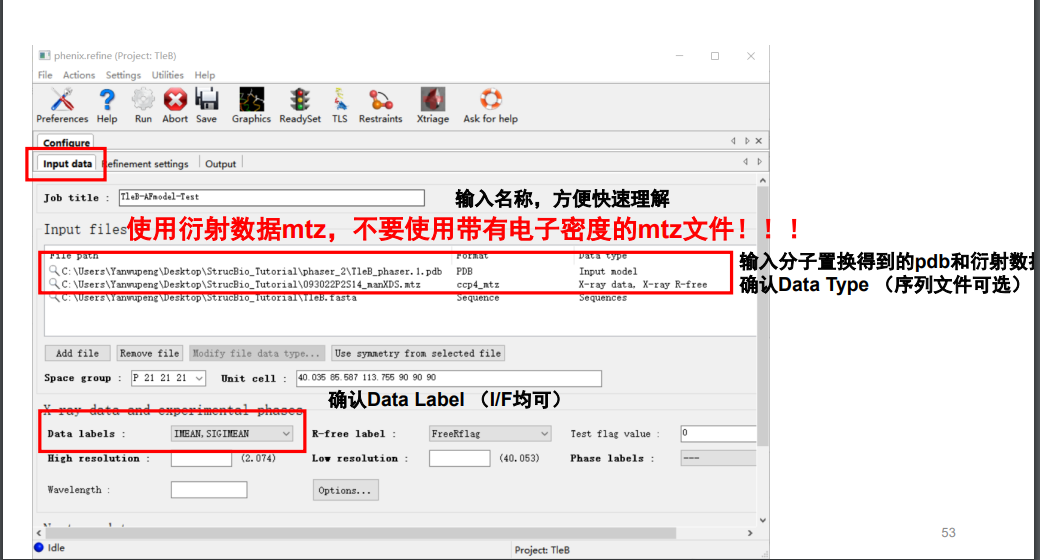
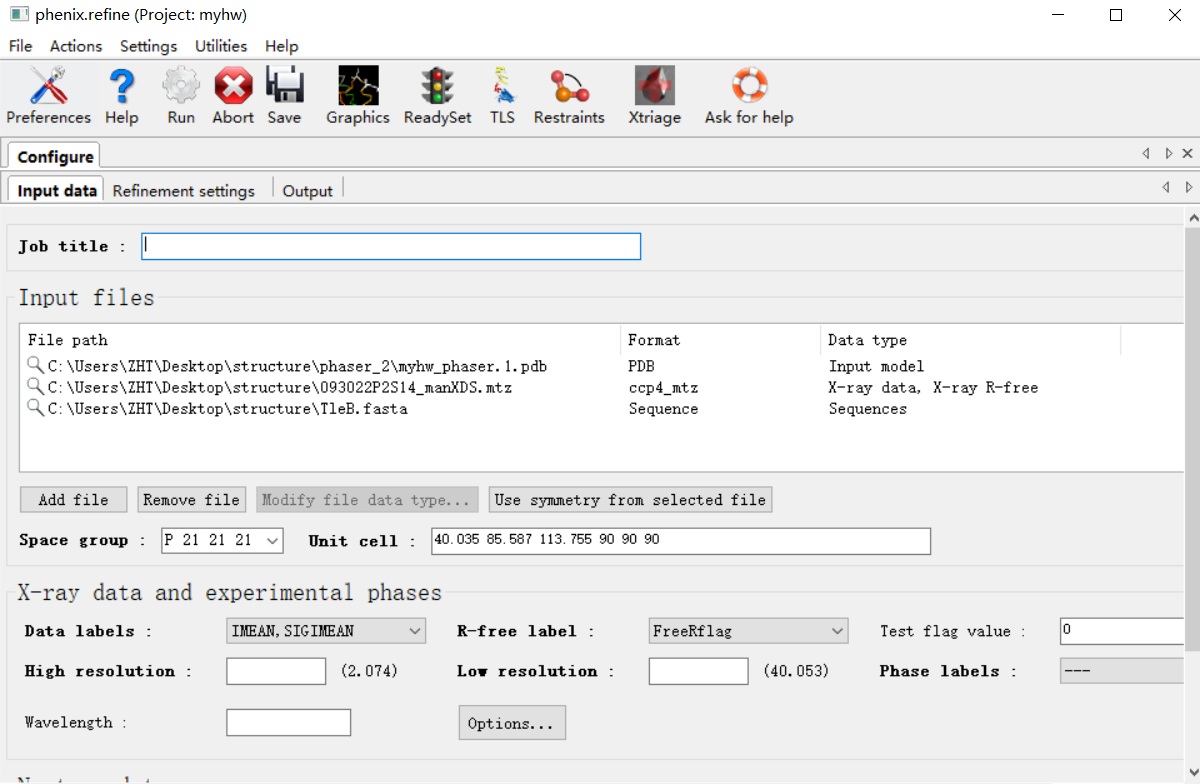
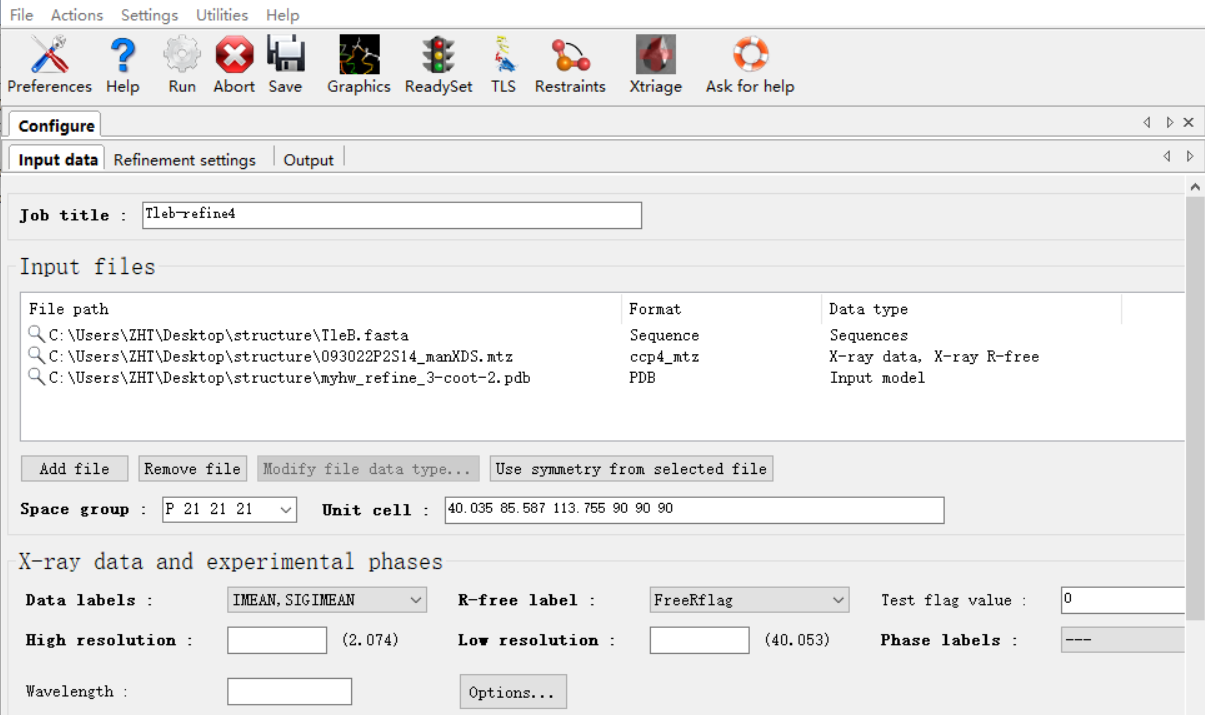
原始的衍射数据(注意!!在精修的时候永远永远使用我们最初的最原始的衍射的mtz的数据),此次的模型pdb,还有序列文件
然后跳到第2栏
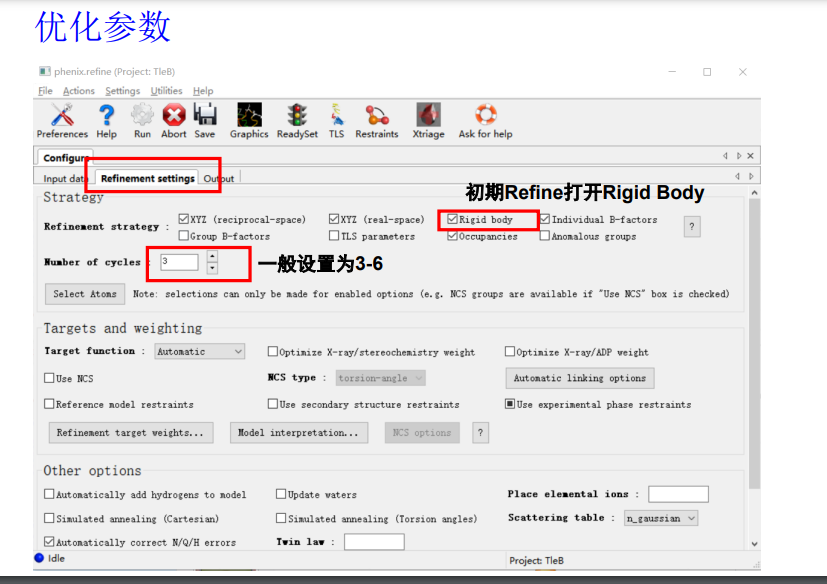
然后注意机器第一遍精修的时候这里只要打开Rigid body(刚性模型)即可,其他的都不要动
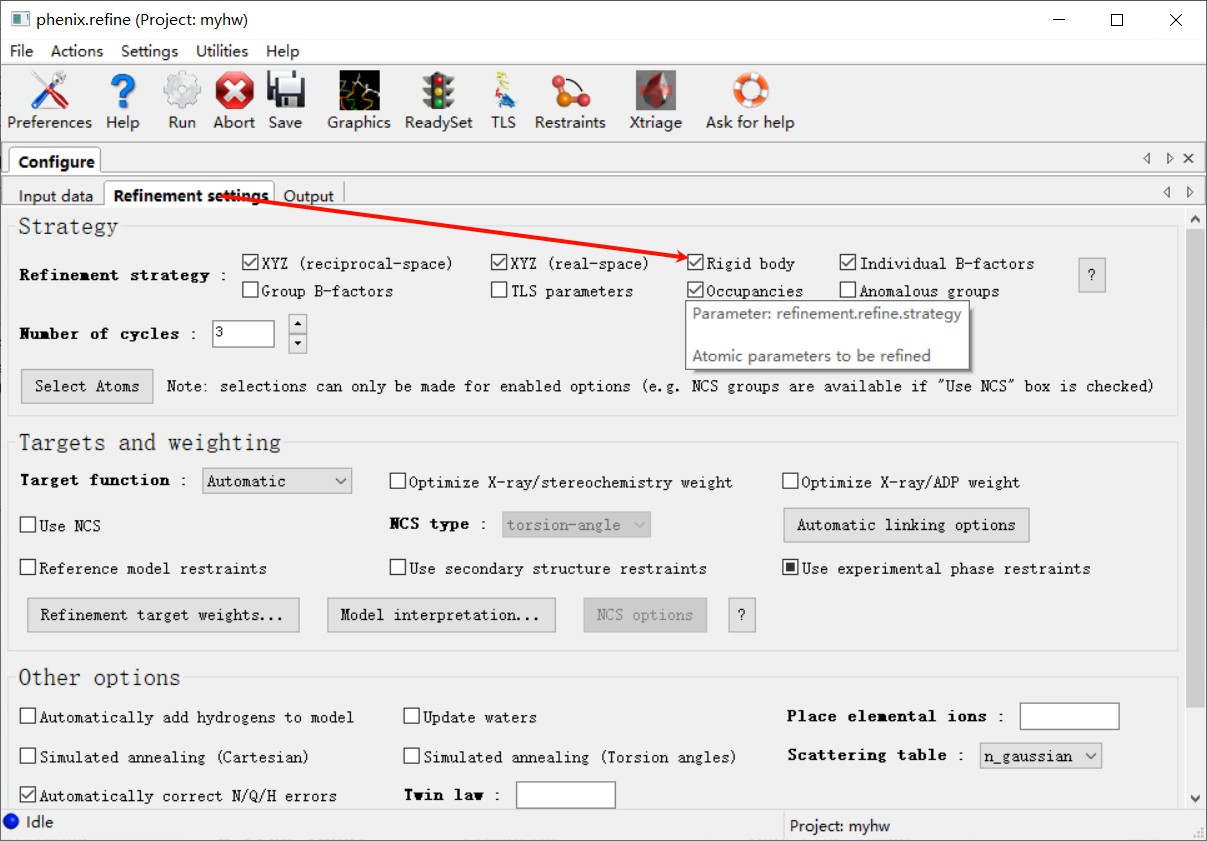
number of cycles一般5轮起步
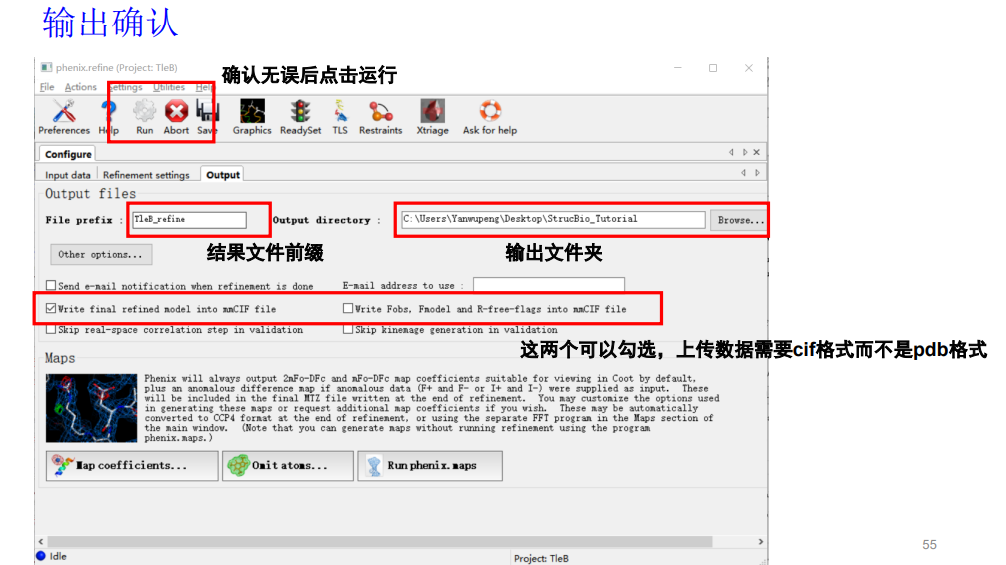
然后就是确认输出的文件位置
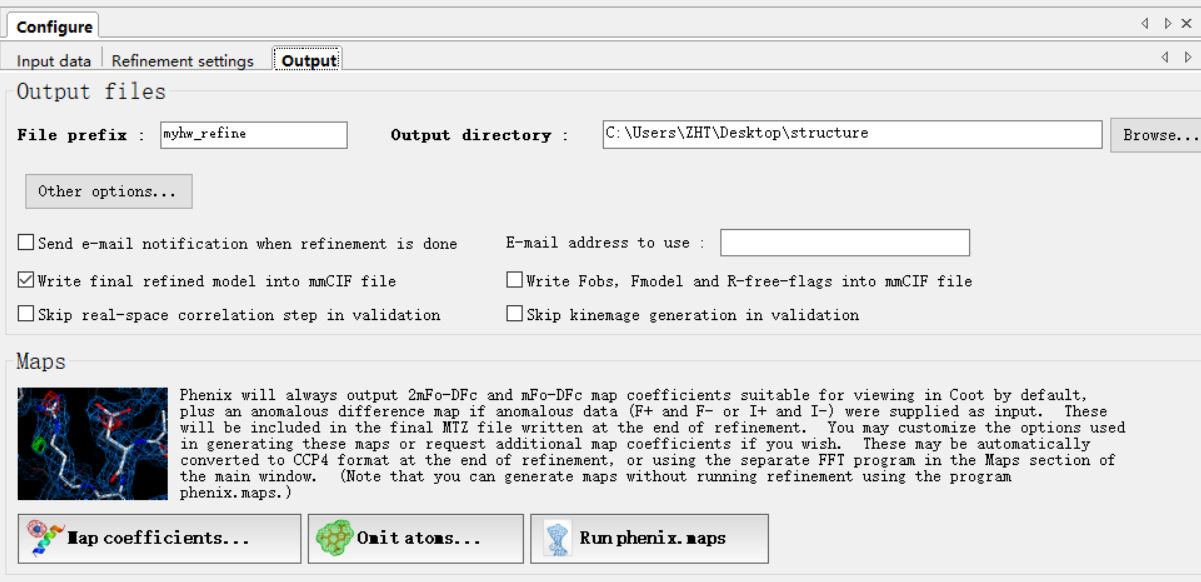
然后就是实时的优化精修进度:
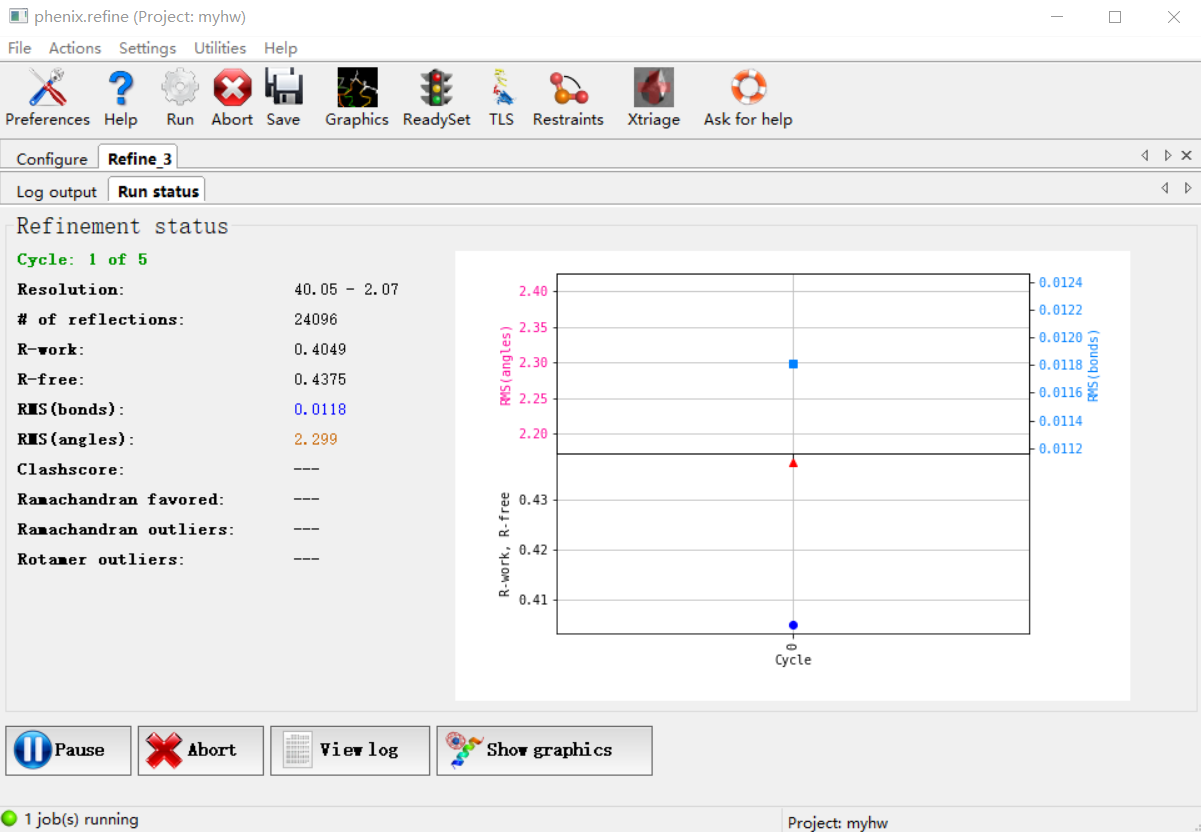
如果关联了coot的话,这一步其实是可以自动打开coot观察精修的过程的
然后跑完之后的结果大概是:
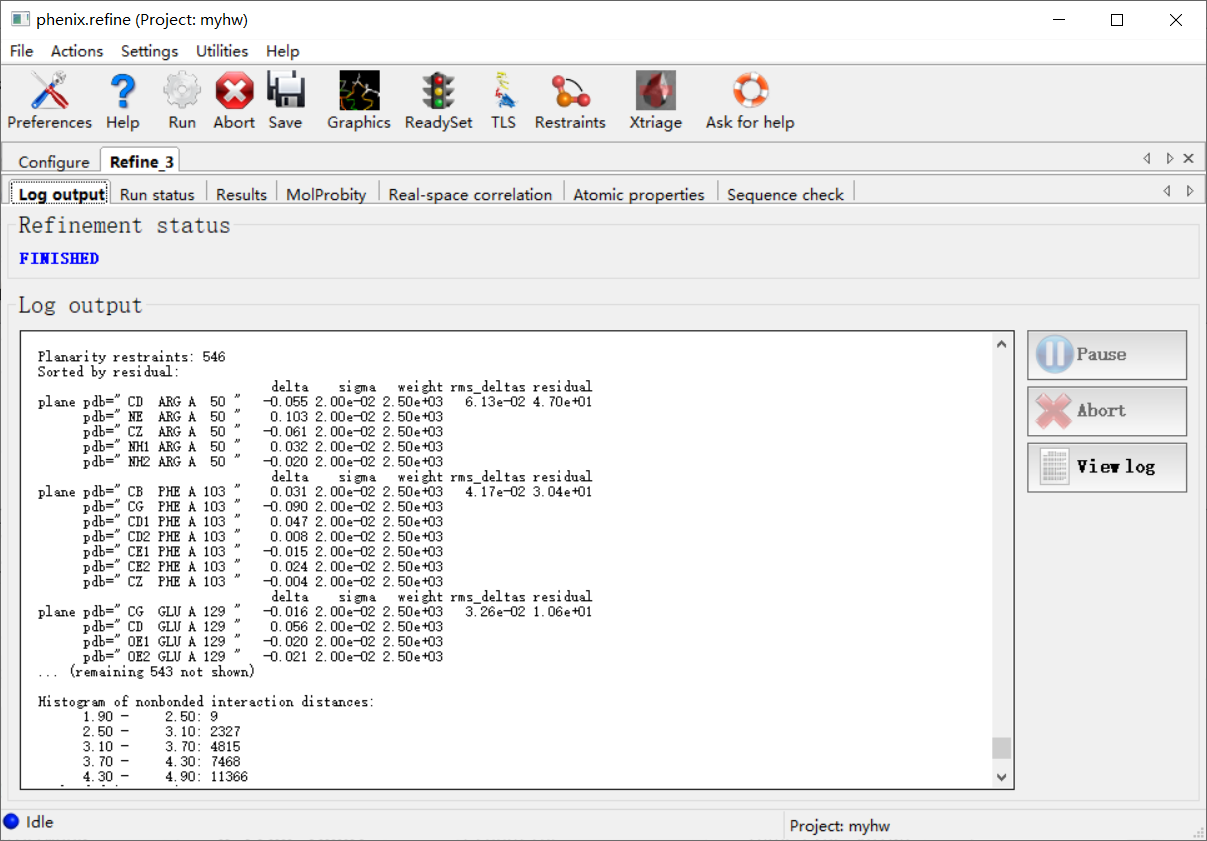
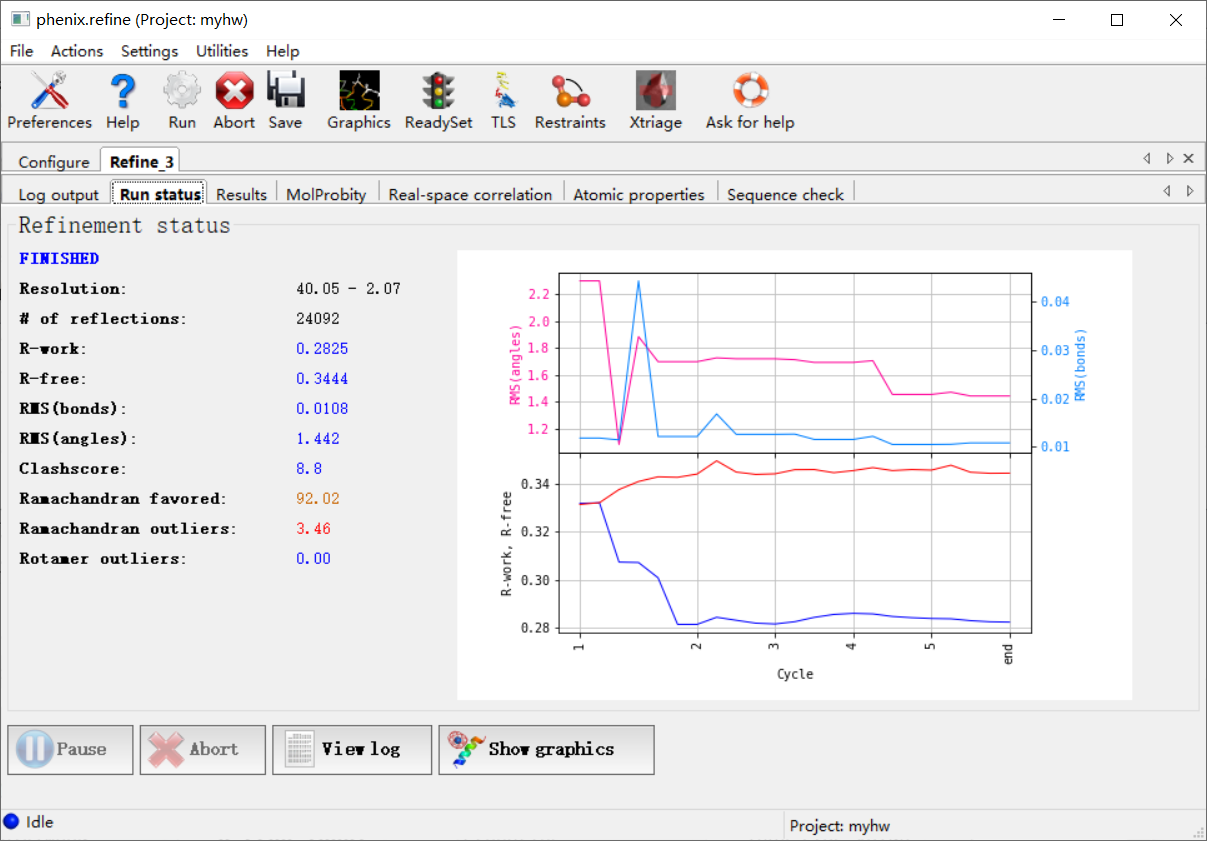
然后左上角这里是全部的输出文件:
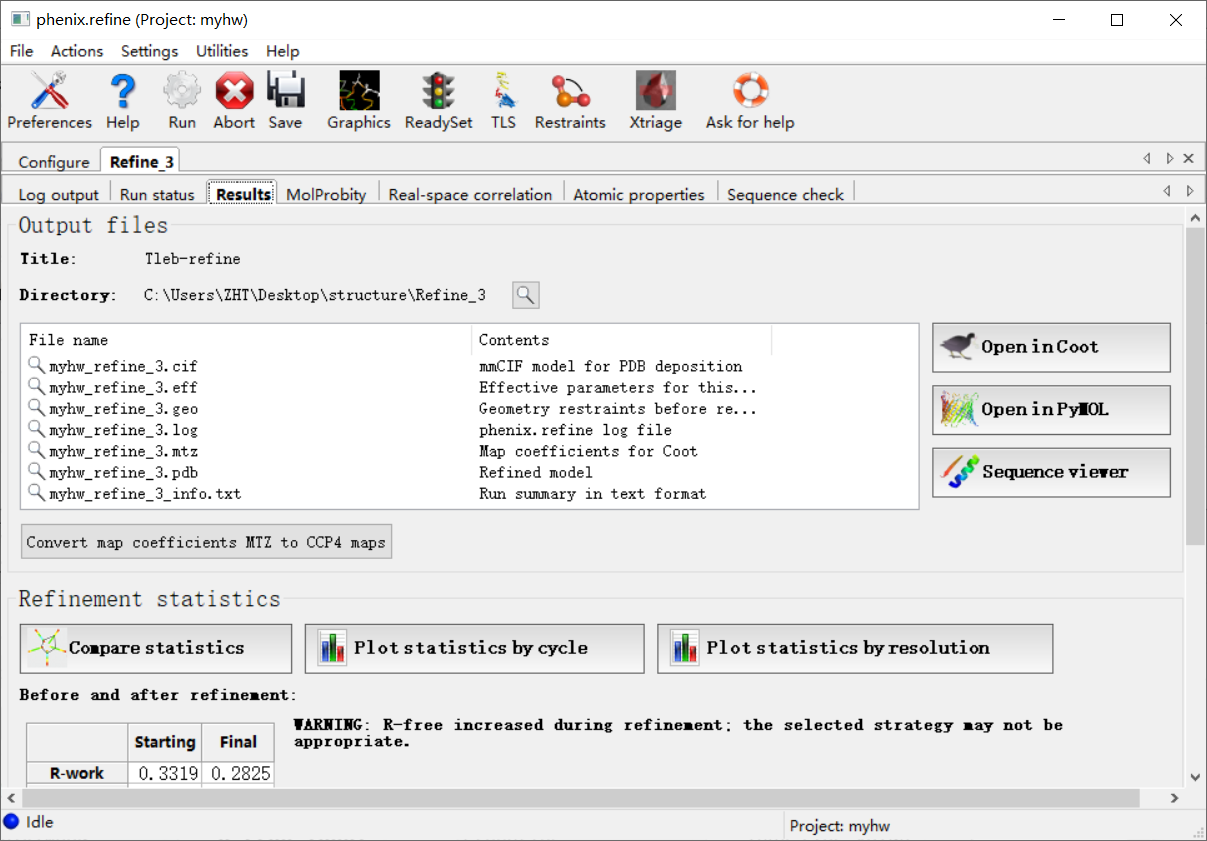
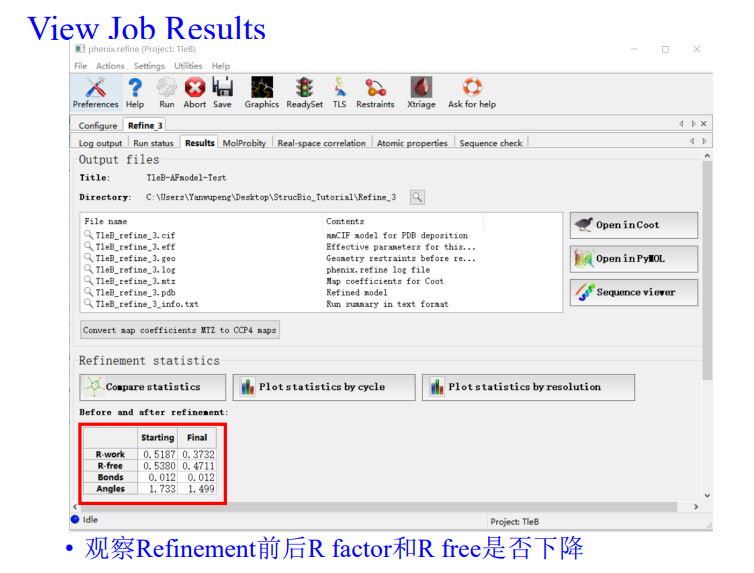
如何评价结果好还是不好,就可以从下面这4个参数中进行判断:
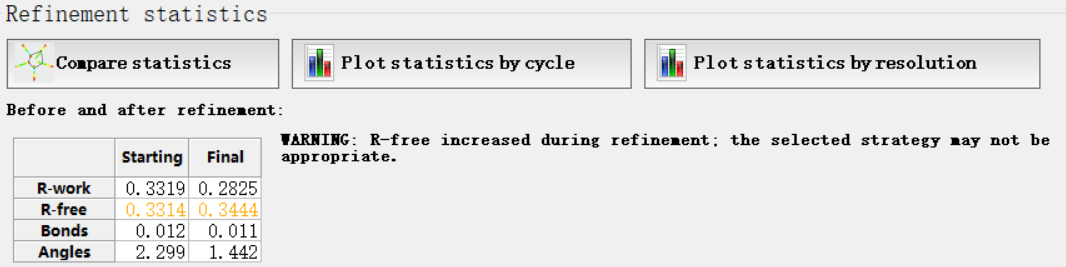
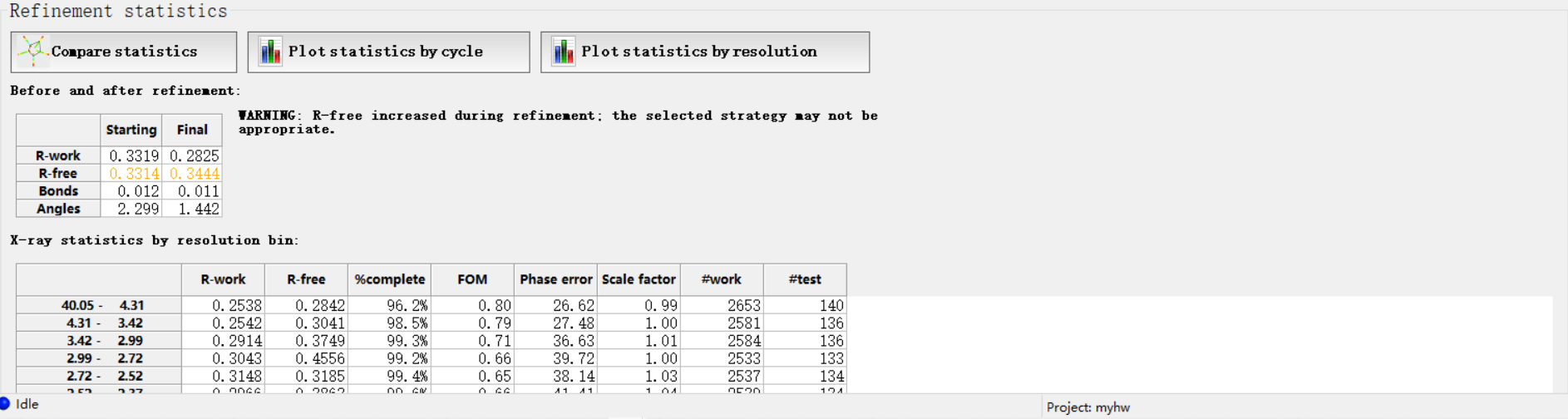
然后这4个指标在指定分辨率范围都有一定的标准值,那我怎么知道呢,其实是可以和标准的进行比较,就是点击下面的
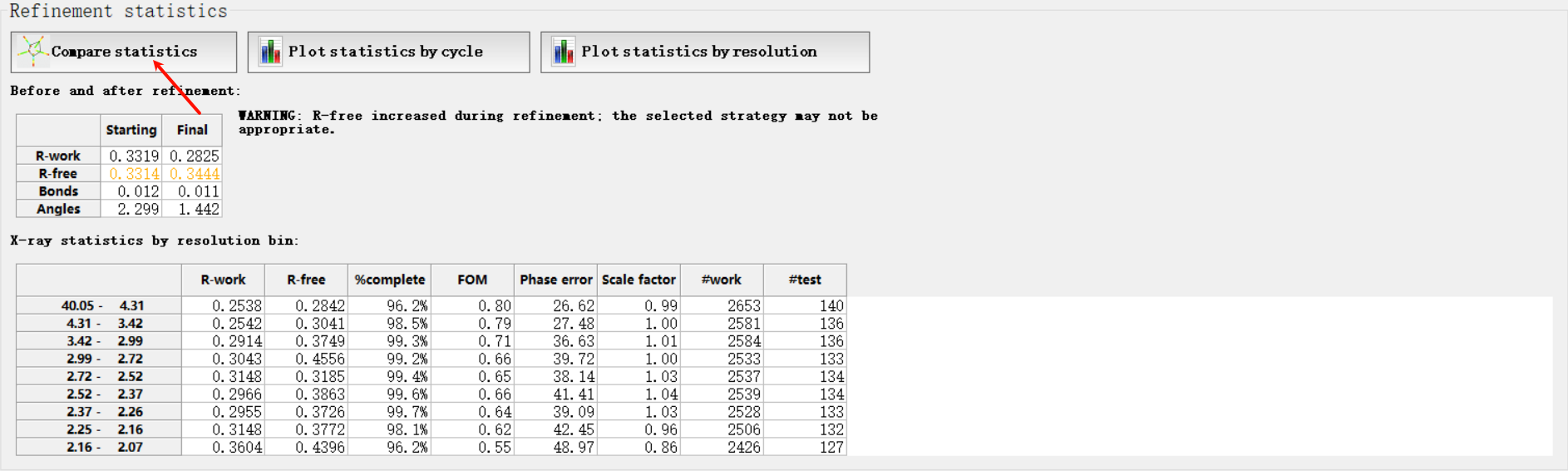
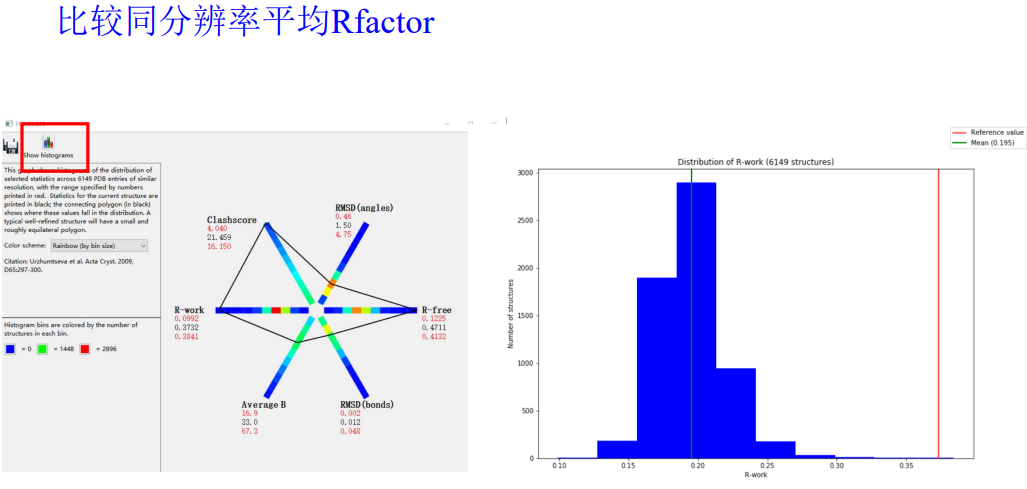
总的来说就是大家的值(大多数分布的地方都在暖色调区域,如果我们在每一个维度坐标上分布的值超过了这些区域范围,就说明我们的分布的正常的标准分布不准)
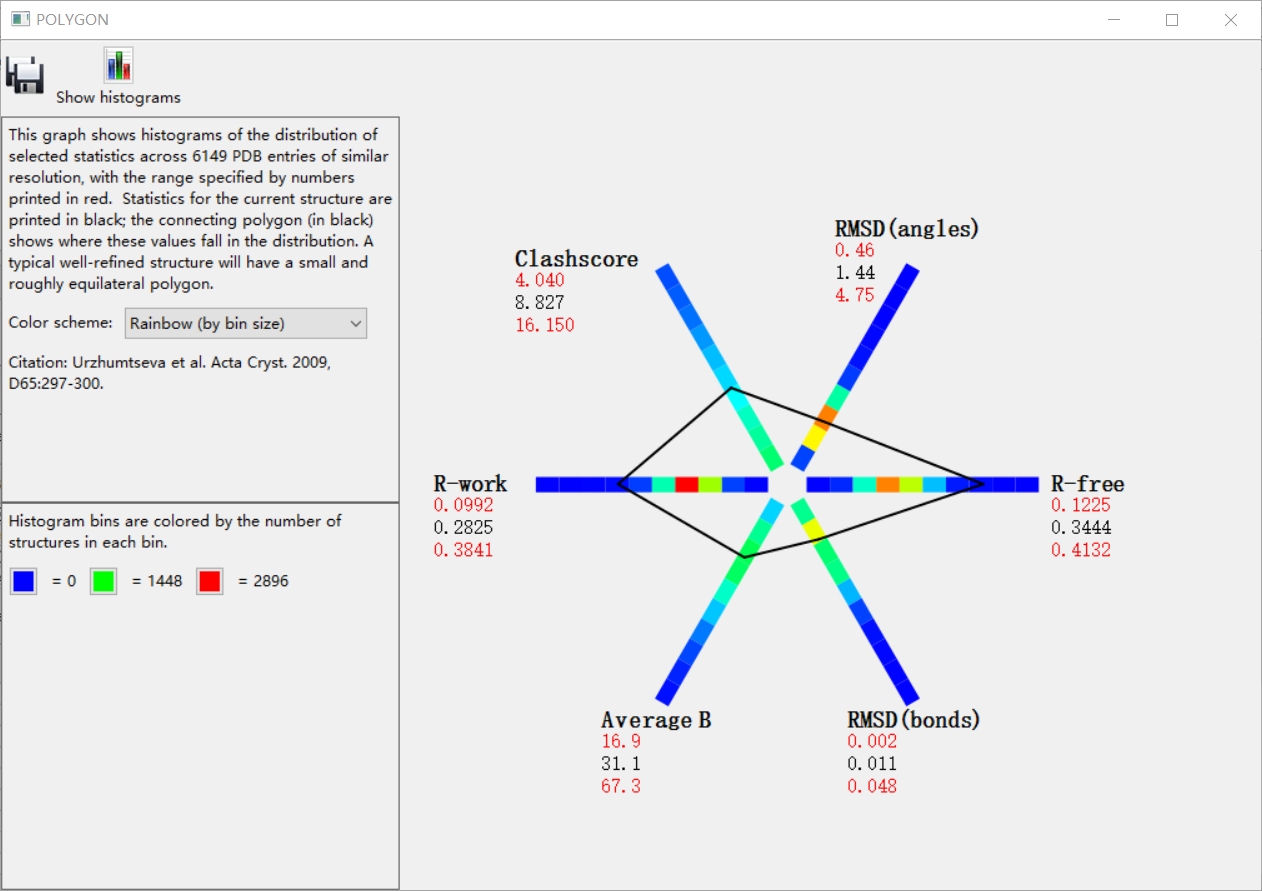
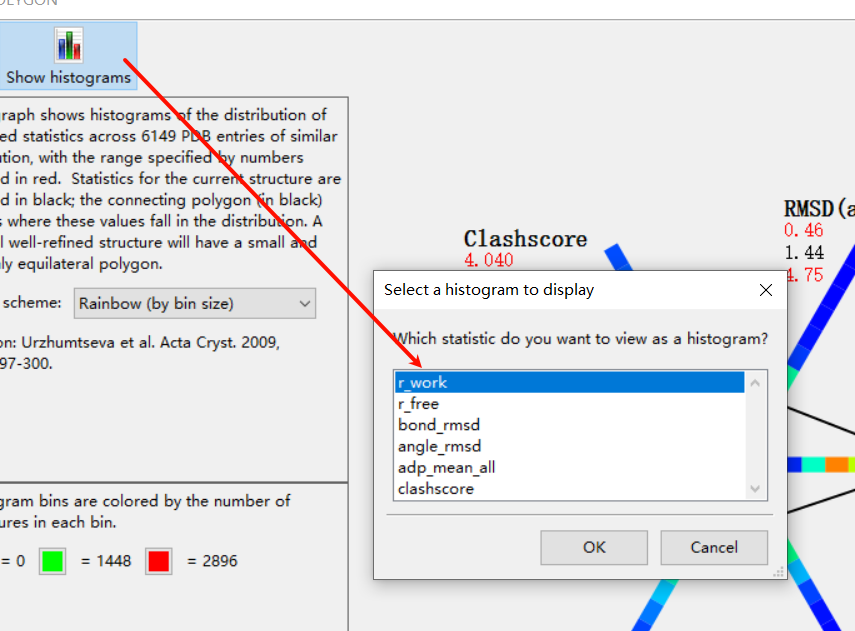
然后每一个维度其实是可以点击上面的直方图展示直接观看效果的:
想看哪个维度就看哪个维度
比如说R-free
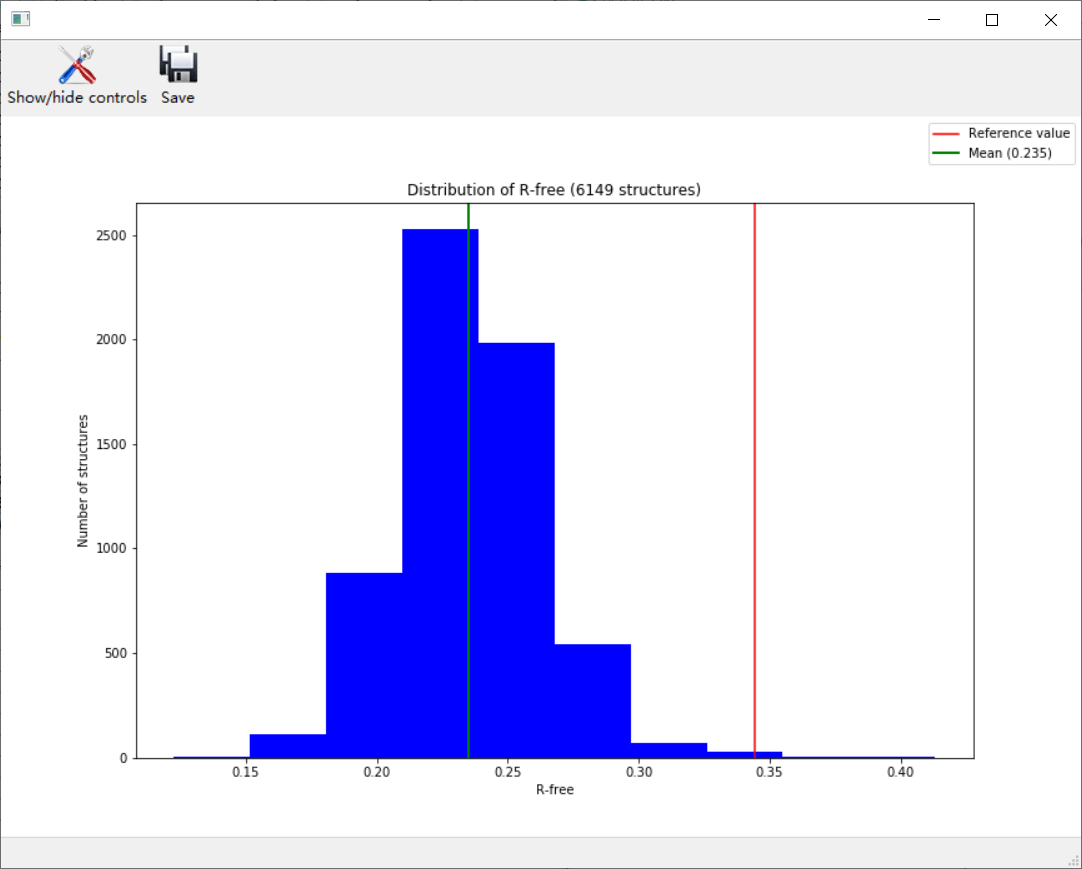
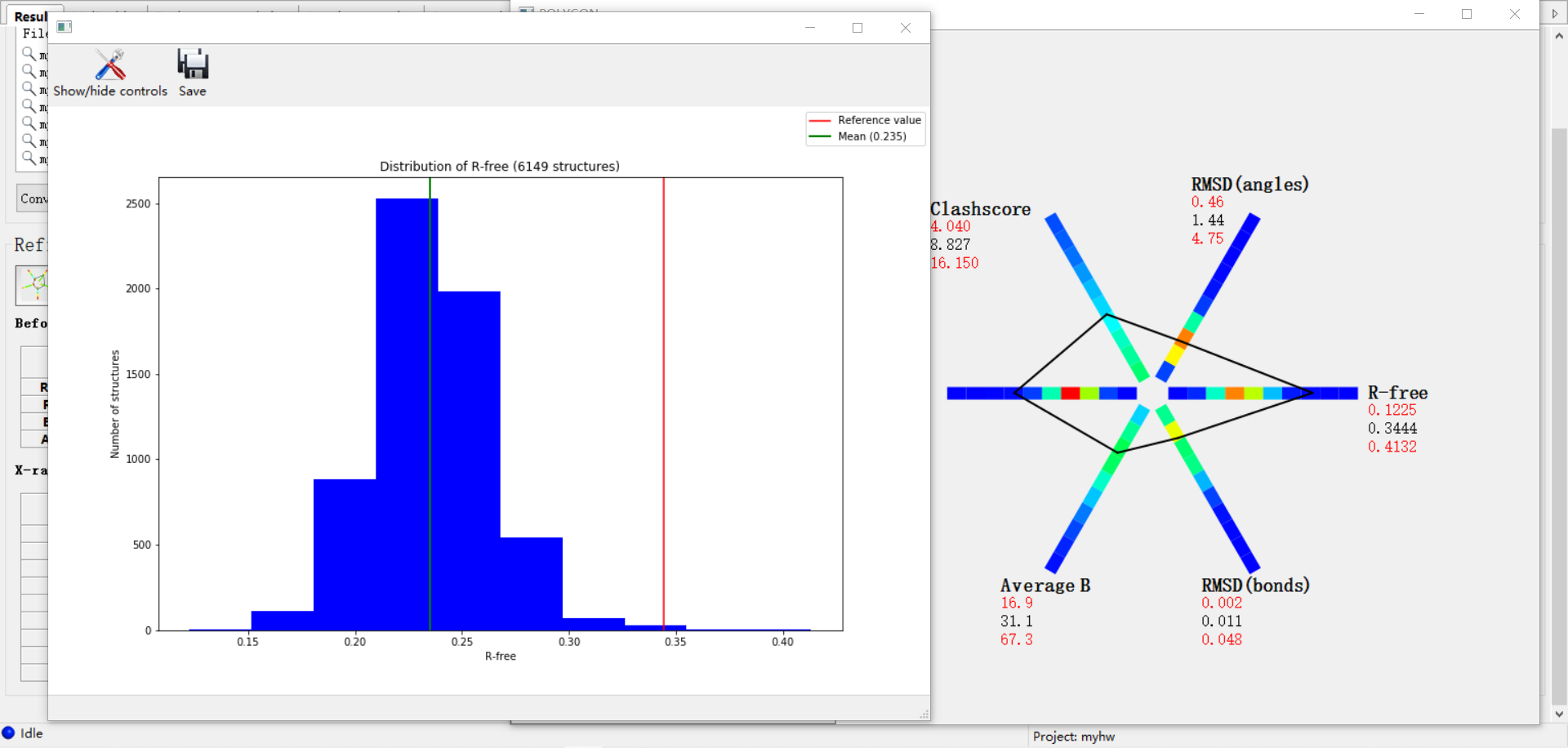
大致上大差不差;
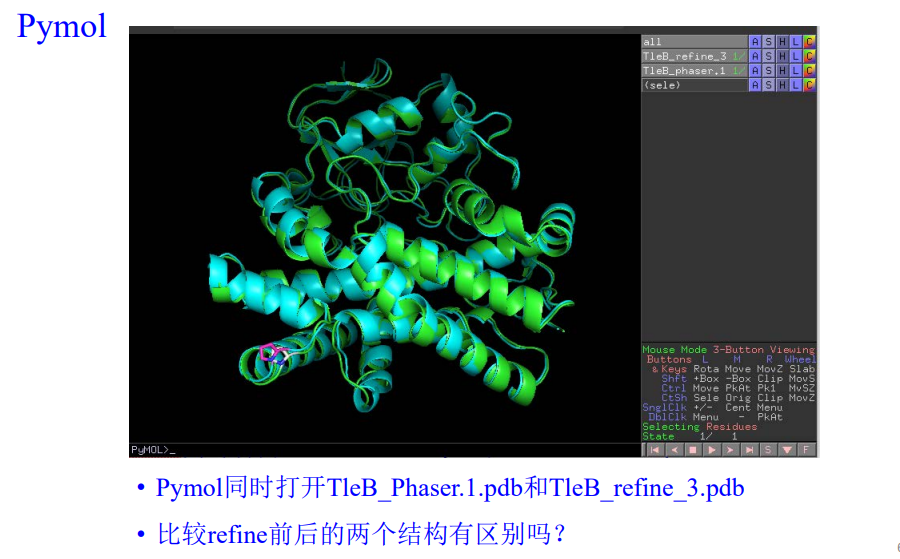
然后同样使用coot查看精修的结果:
注意结果是在这里:refine3的mtz以及pdb
其实可以看到:在精修之后出现了很多红色以及绿色的电子云
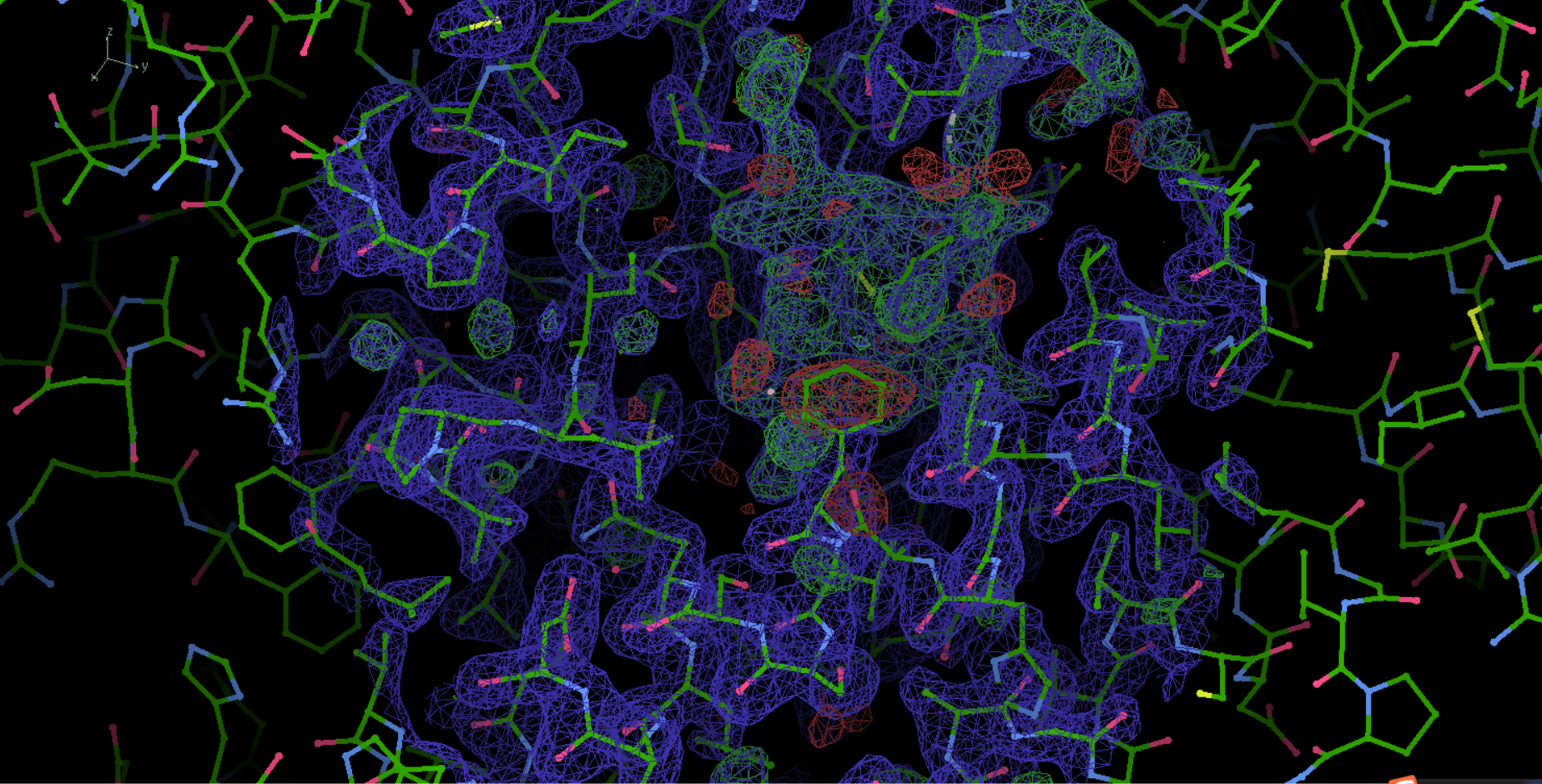
=====》过程中如果不小心点击退出去程序了,可以重新打开
然后在操作历史里找到对应的操作执行部分即可,比如说上面是refine部分,我们就可以直接点击返回到之前的结果部分了
6,在coot中进行第一次人工精修:
将4个选项都选上
第一次人工精修的话,最好是从头到尾,也就是从蛋白的N端到C端;
可以goto选项,
或者点击空格键,就是跳到第一个原子,然后接着按空格就是前进,如果是shift+空格就是后退
可以放大看细节编号:
然后我这里的模型是最小只能跳到7号:
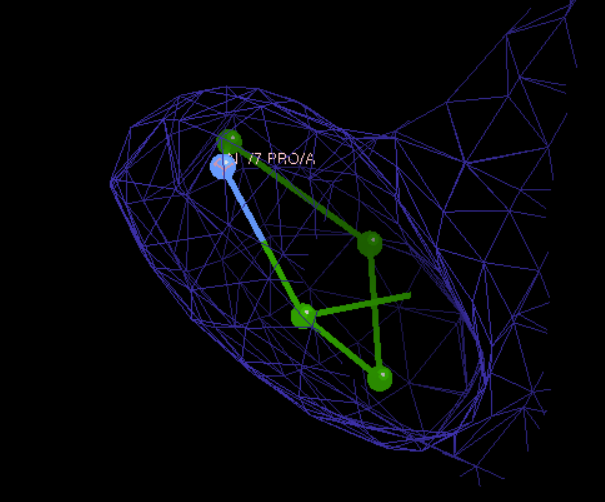
然后一个一个看的话,比如说我们可以看到12号这里有一个红色电子云
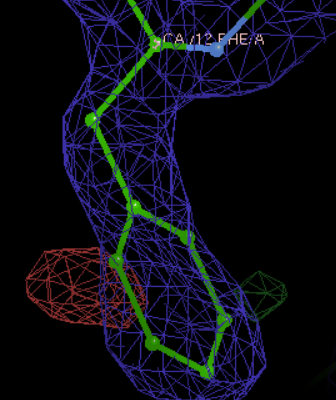
然后景深以及map level等需要进一步调整一下参数,在修改的时候可以更方便。
(1)然后可以对照序列进行分析:
打开一开始机器精修的refine部分的sequence check部分
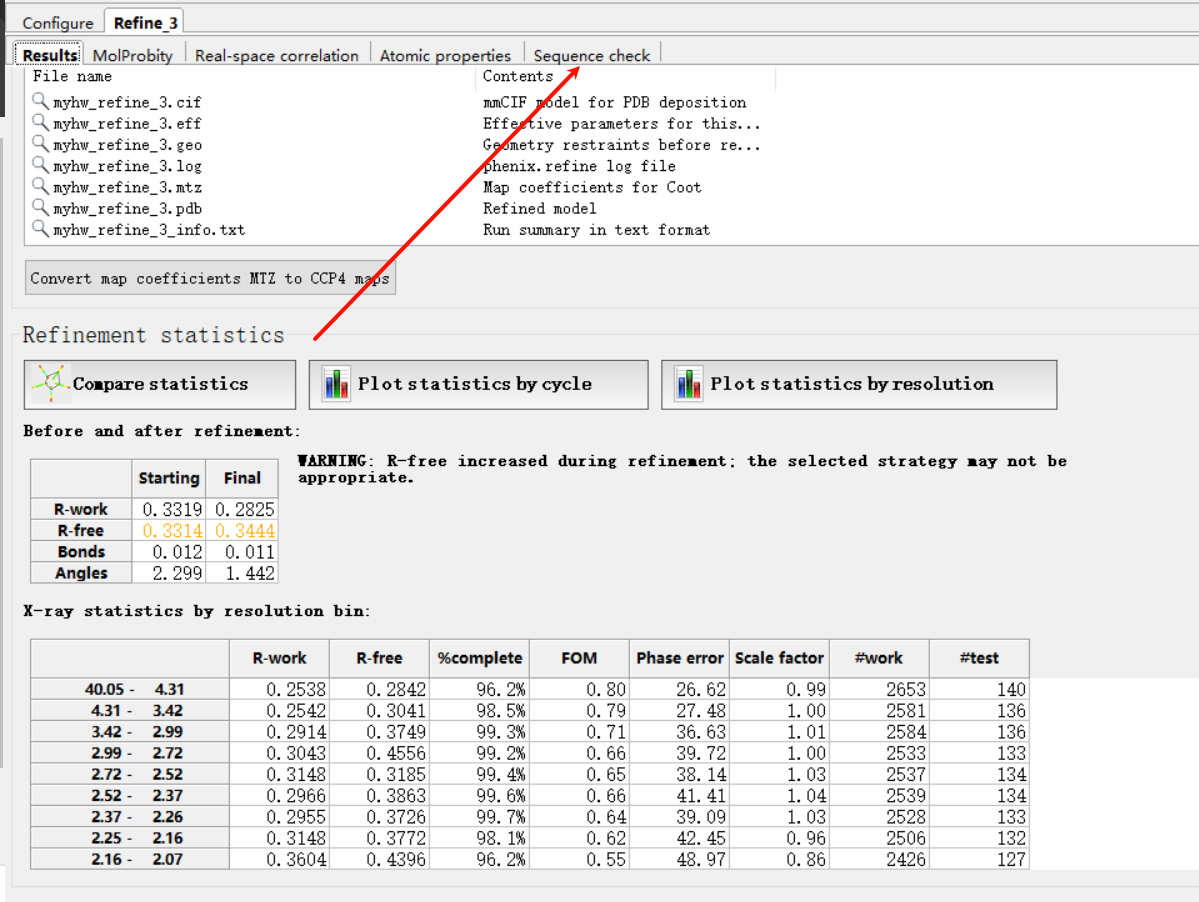
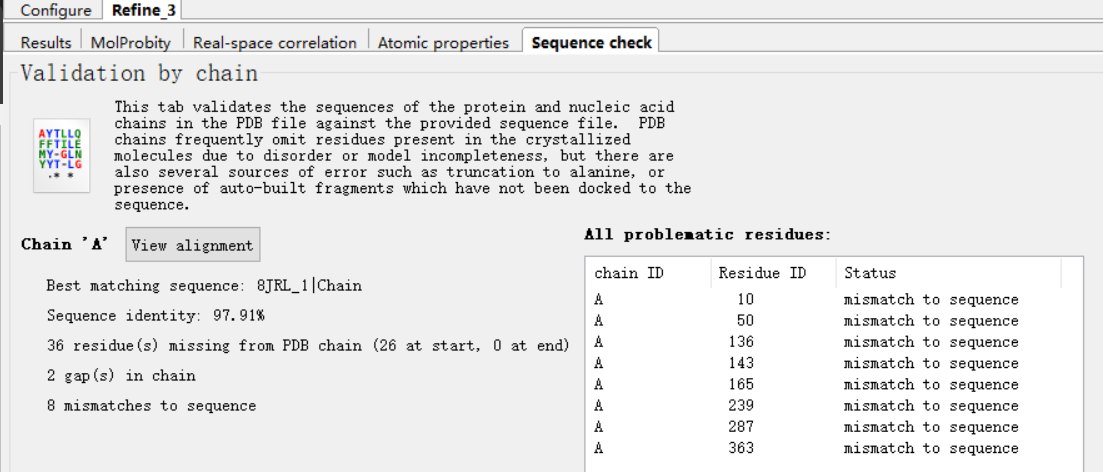
比如说这里说10号位有一个mismatch
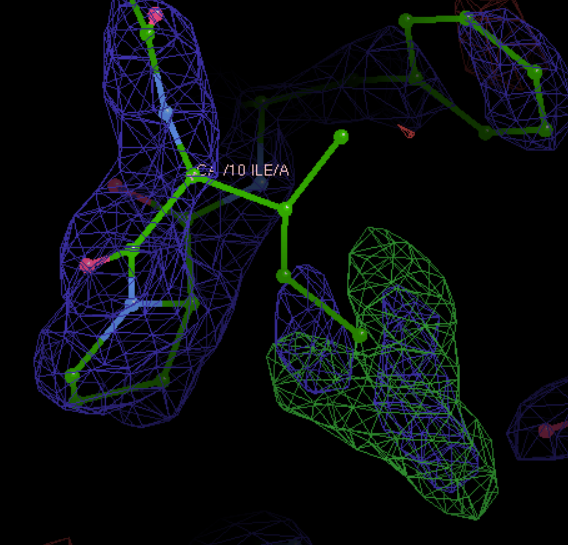
其实可以通过往前往后跳动观察一下是否有红绿色的电子云到底靠近编号是几的C-α,然后就可以判断到底是哪个位置上的氨基酸是有问题的了;总之从上面结果看,应该是10号位的氨基酸的侧链上的的电子云密度有点问题,当然对于氨基酸来说,侧链有问题,基本上就自然会想到侧链obs与模型不一致,而自然的氨基酸实际上也就是在侧链上不一样,所以很自然就想到是不是这里的氨基酸建模建错了,比如说应该是另外一个氨基酸;
总之我们接着查看sequence check部分:
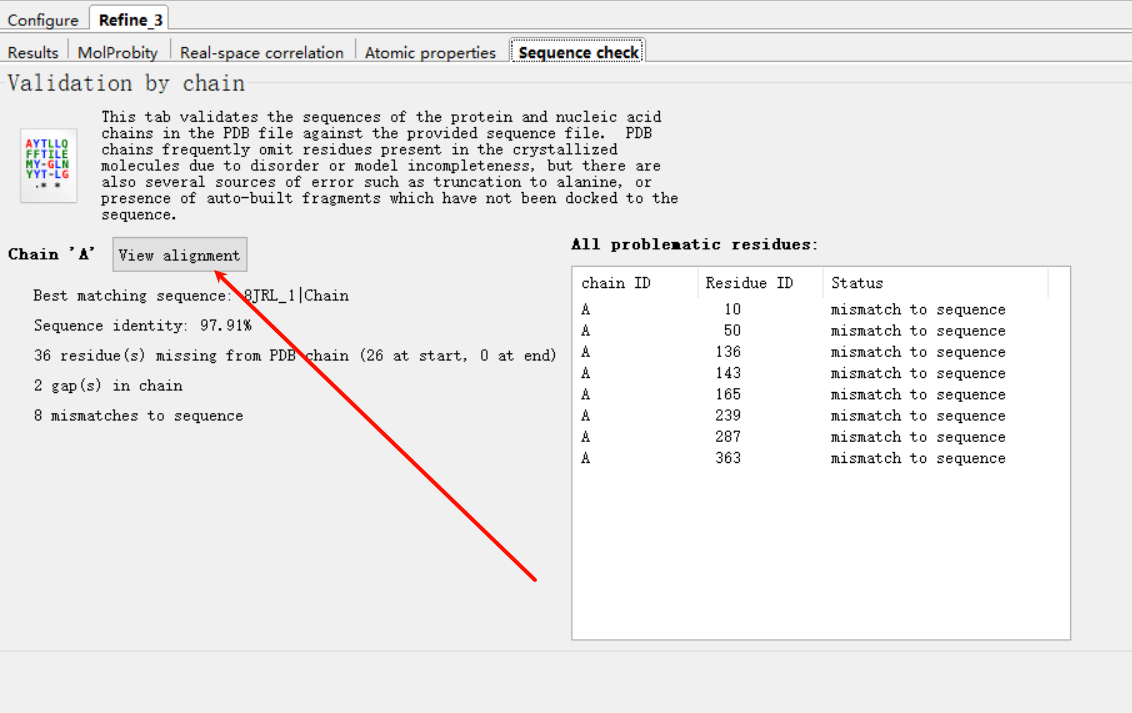
细节可以view alignment

因为我们是从7开始,789-10,正好是第10个,
然后我们这里是Ile异亮氨酸
正常应该是Y也就是Tyr酪氨酸
那么我们精修的时候应该如何修改呢,点击右边的这个按钮:
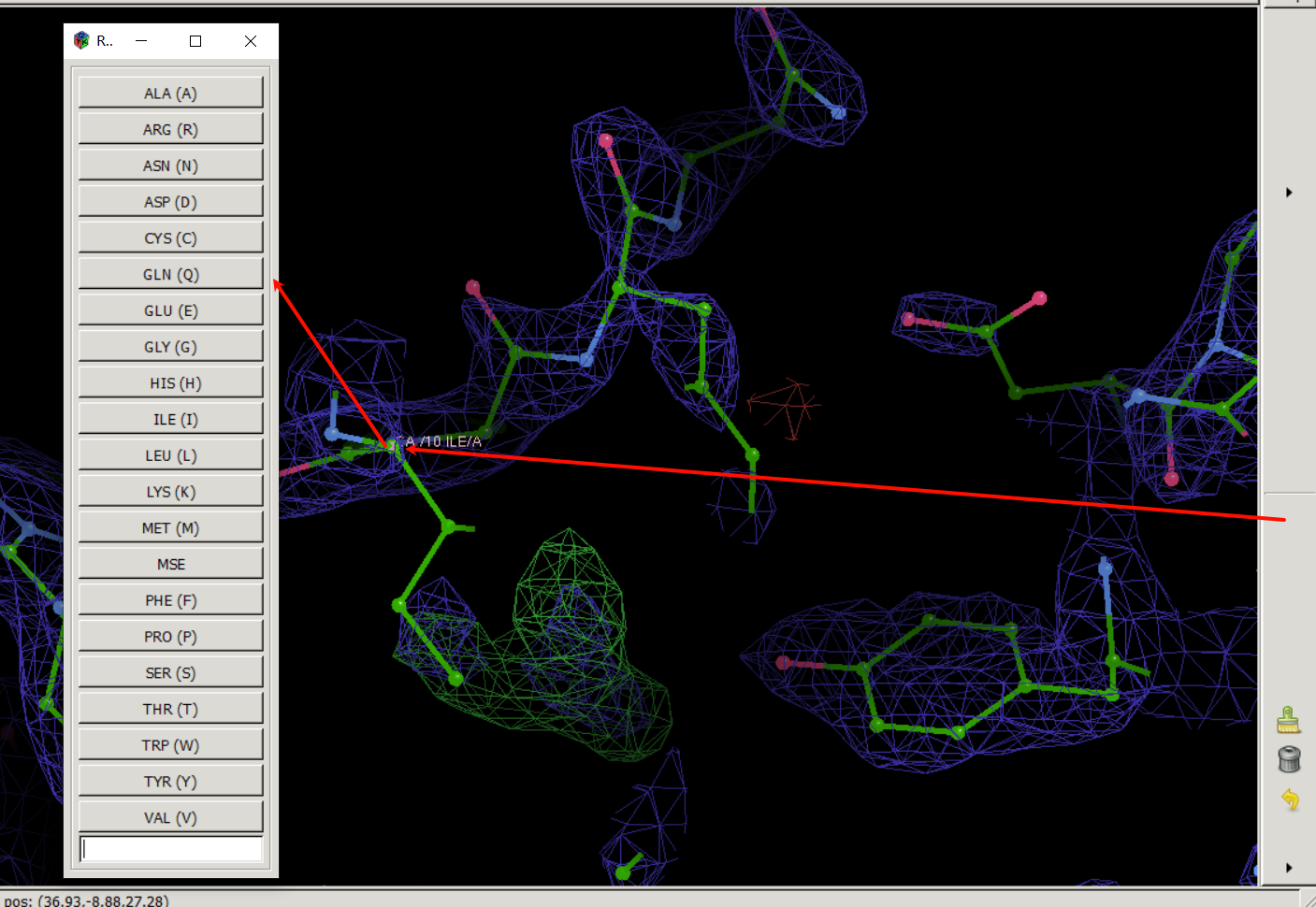
我们点击这个按钮之后再点击我们要修改的10号位的氨基酸残基上的任意一个原子(都会被识别为要修改该号位的氨基酸)
然后就会弹出来一个要修改的列表
点击修改为Y即可
——》此处发现windows上的wincoot没安装好,换了一个linux上的coot接着按照上面操作
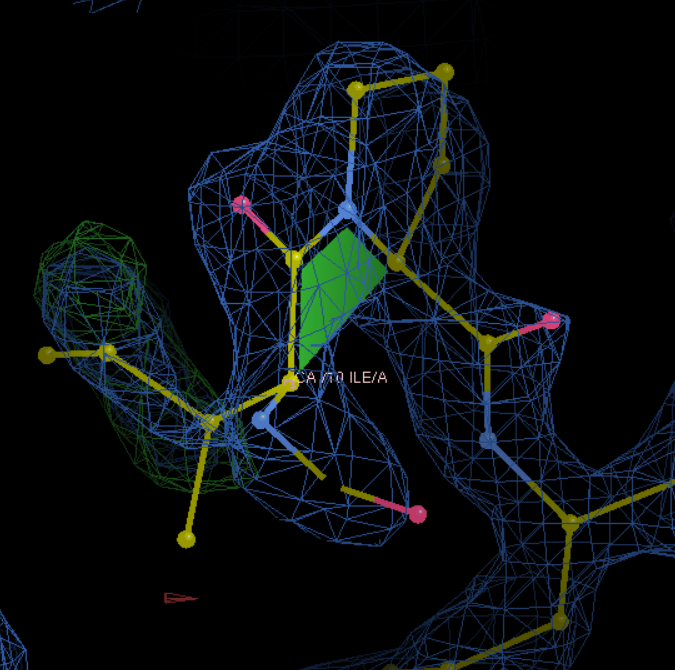
总之就是10这里不对,需要修改
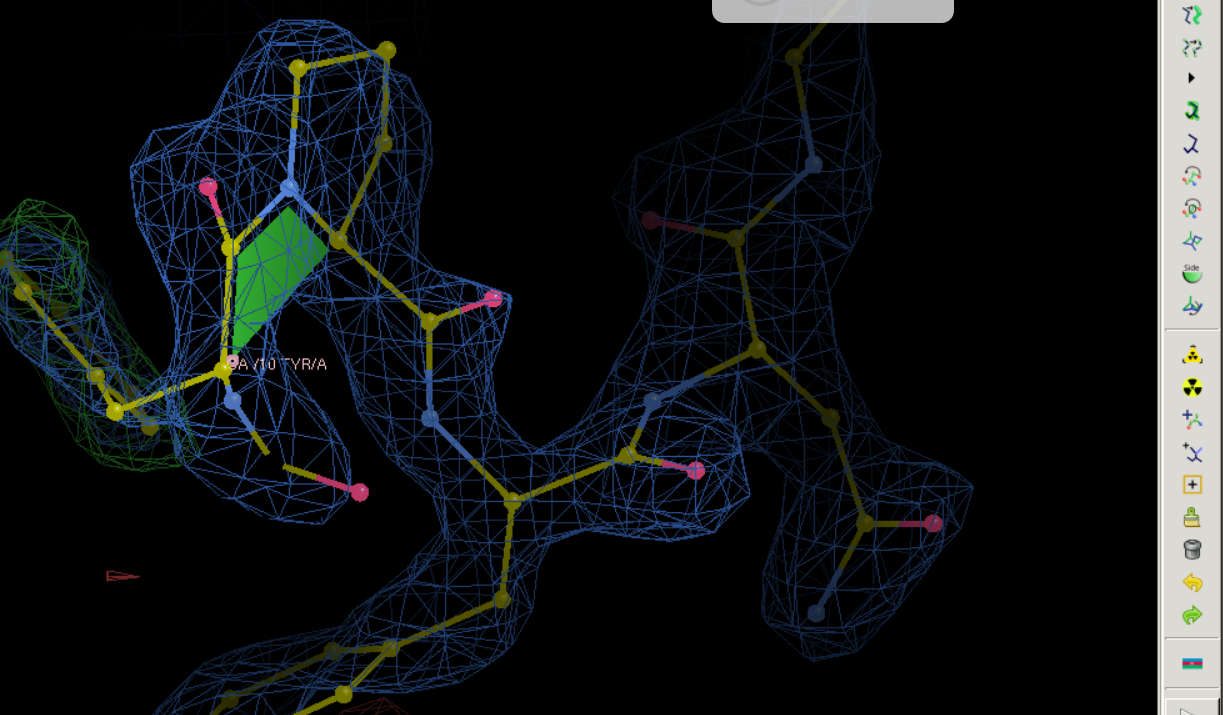
果然,在linux上修改效果比较好,变成了Tyr
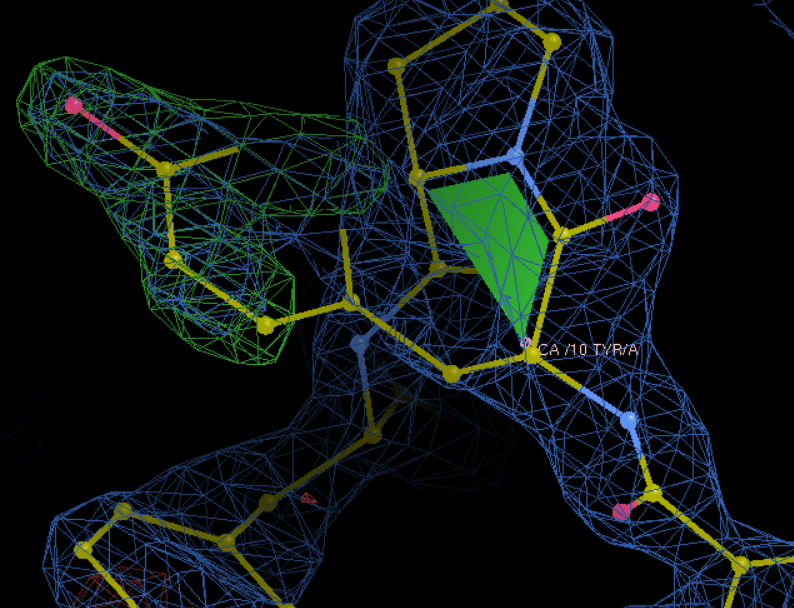
总之就是按照这里的进行修改:
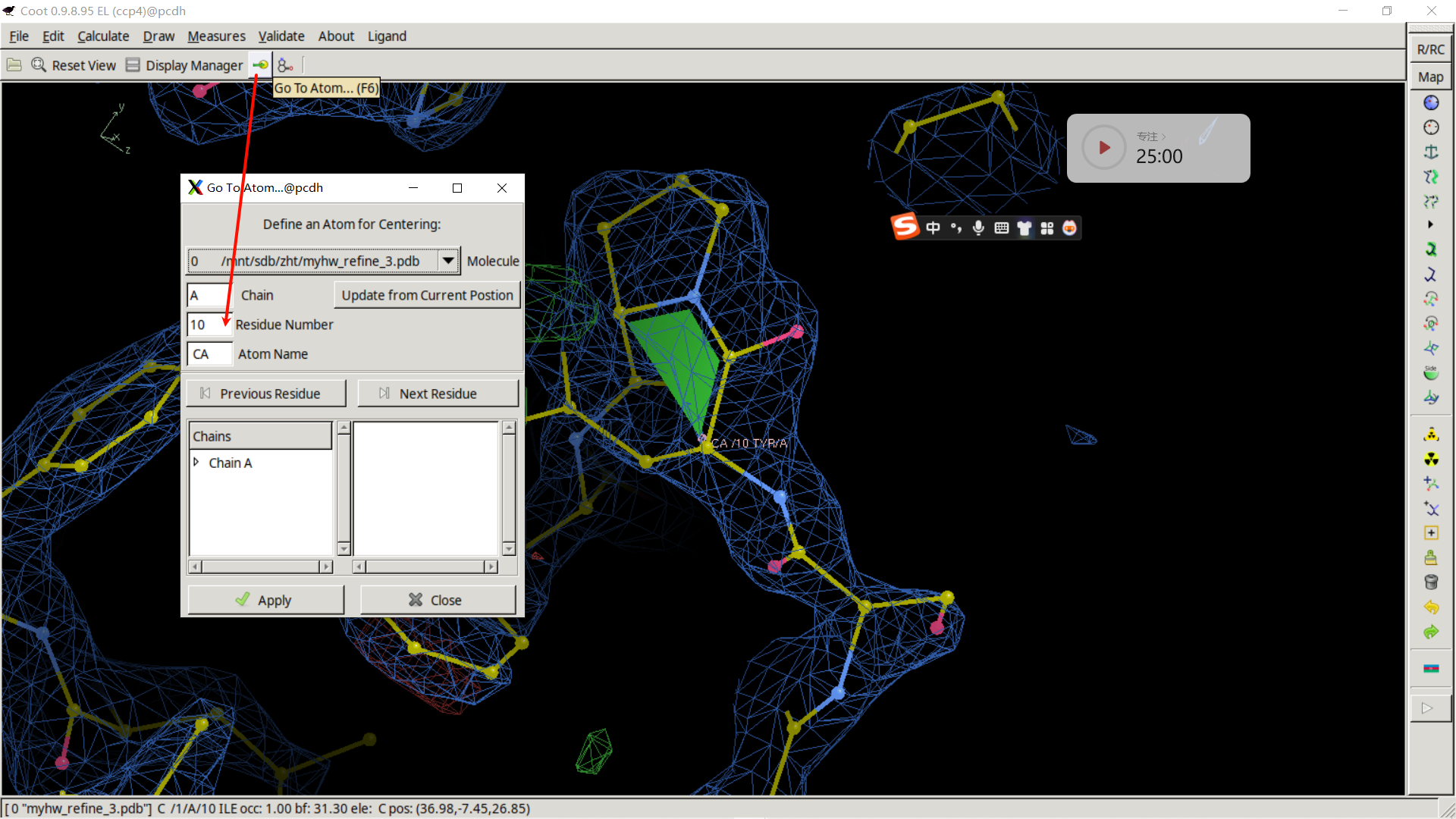
直接这里就有goto哪个哪个atom
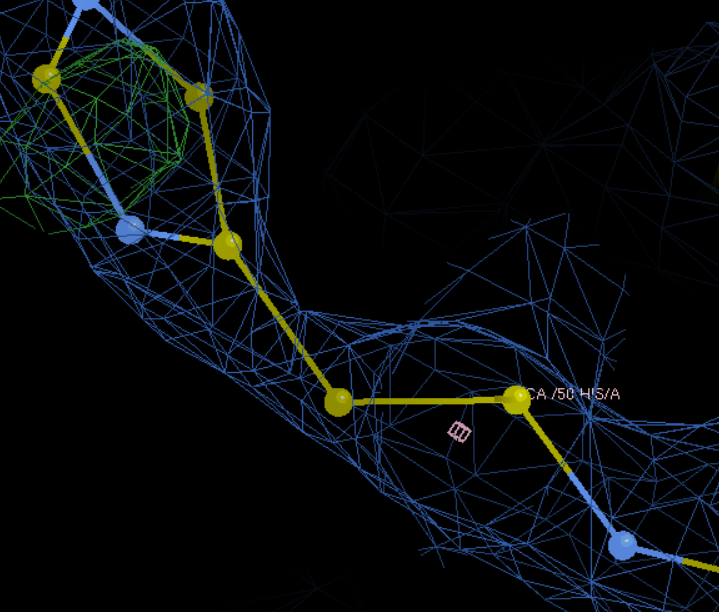
下一个就是50,接着修改:
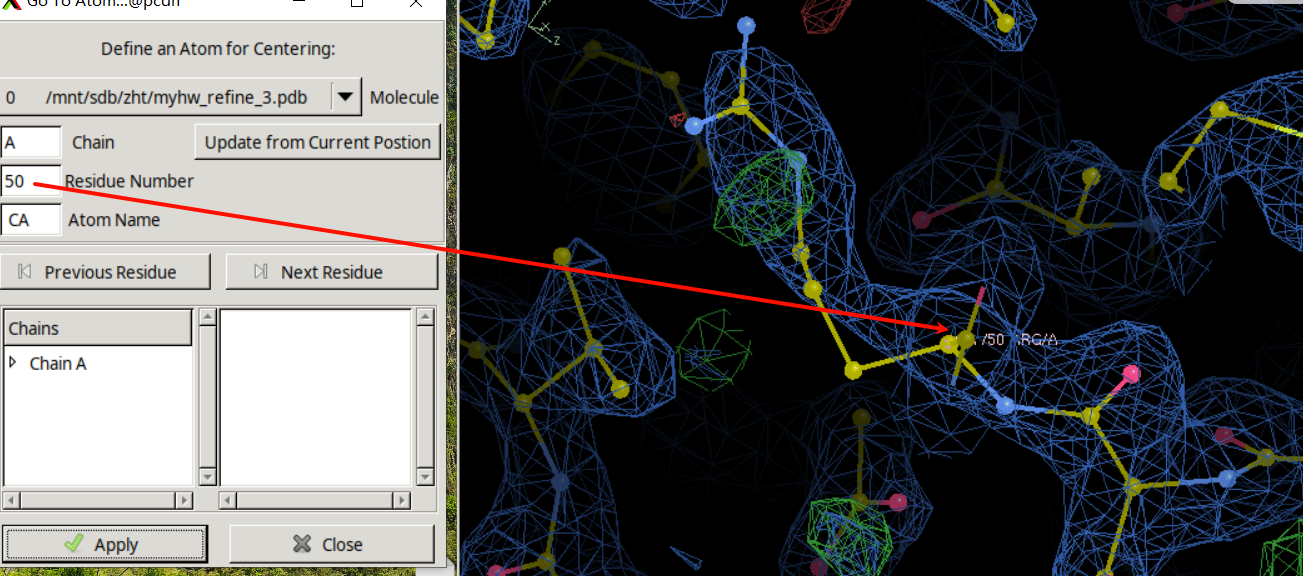
R改成H
同样改完其他的氨基酸残基:
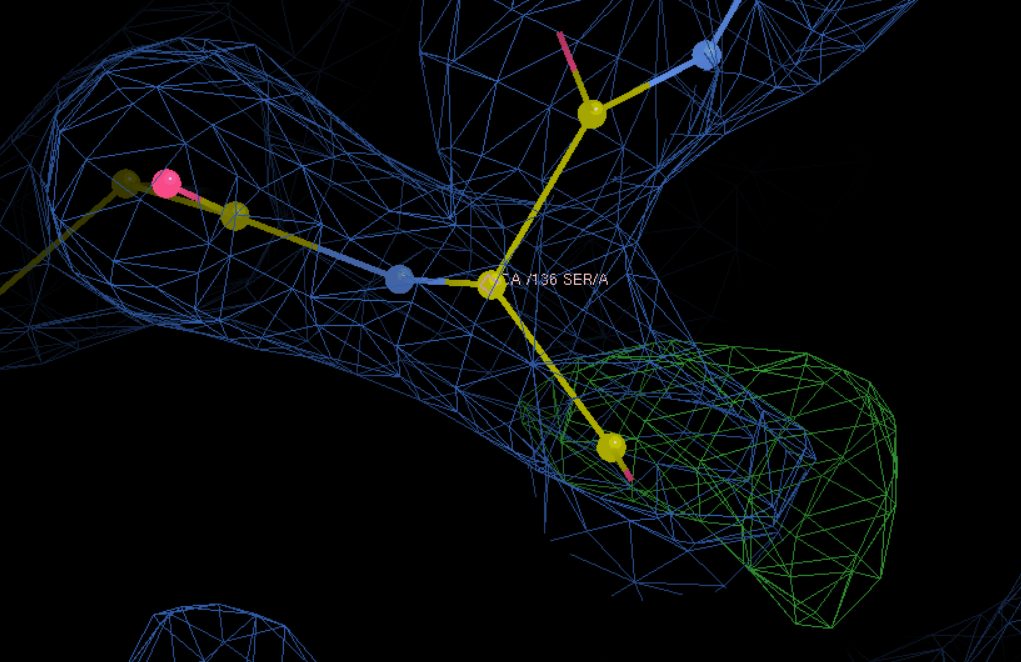
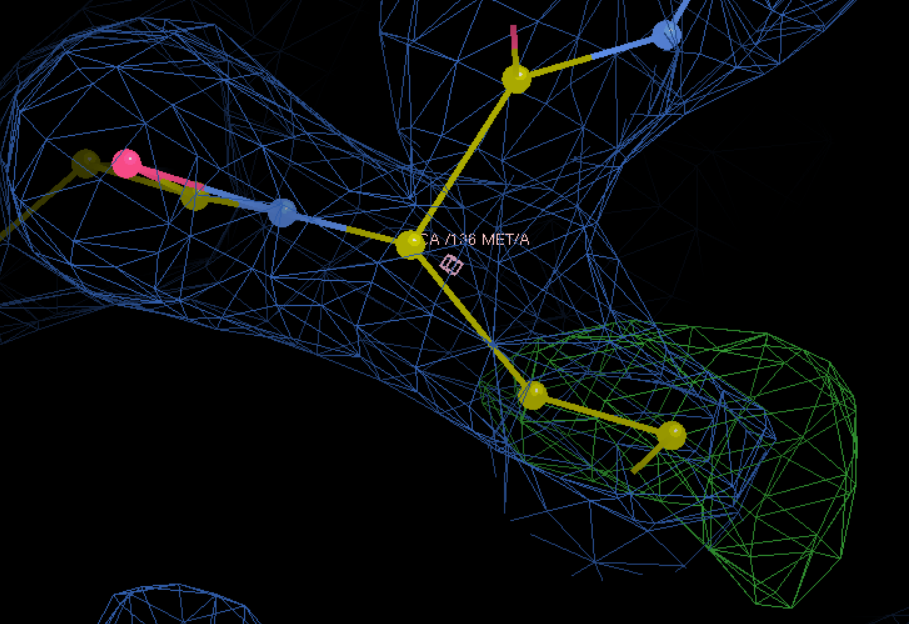
S改为M,再接着
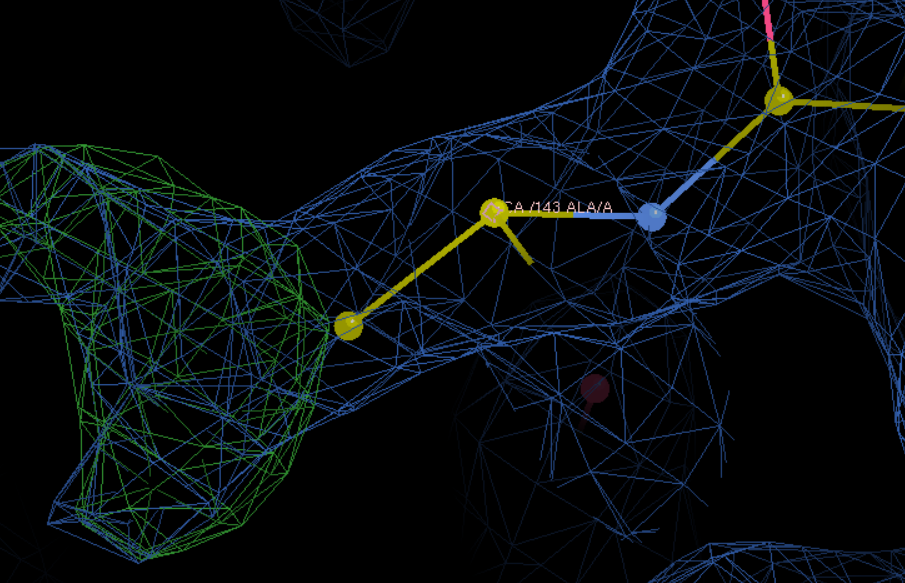
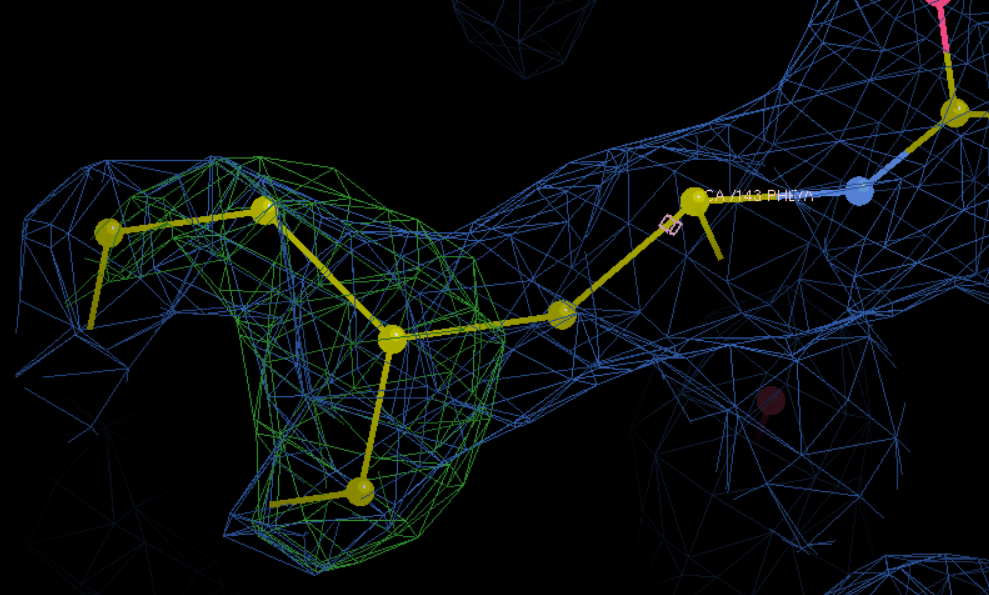
A改为F
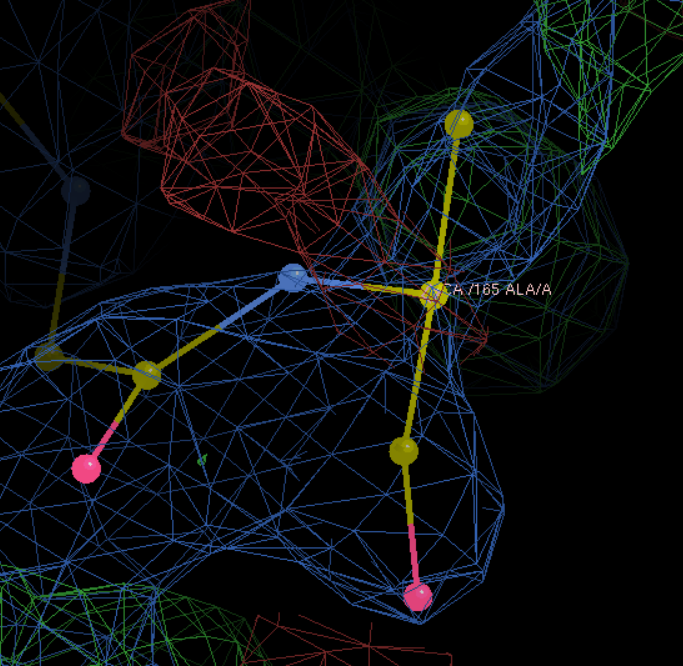
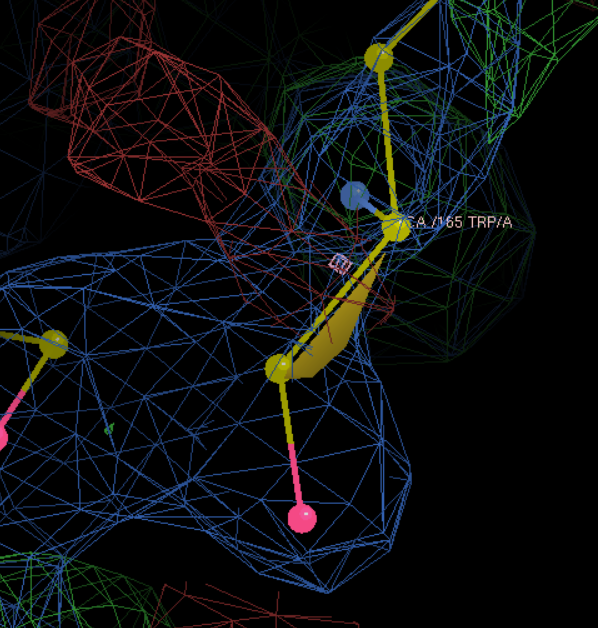
A改为W
F改为T
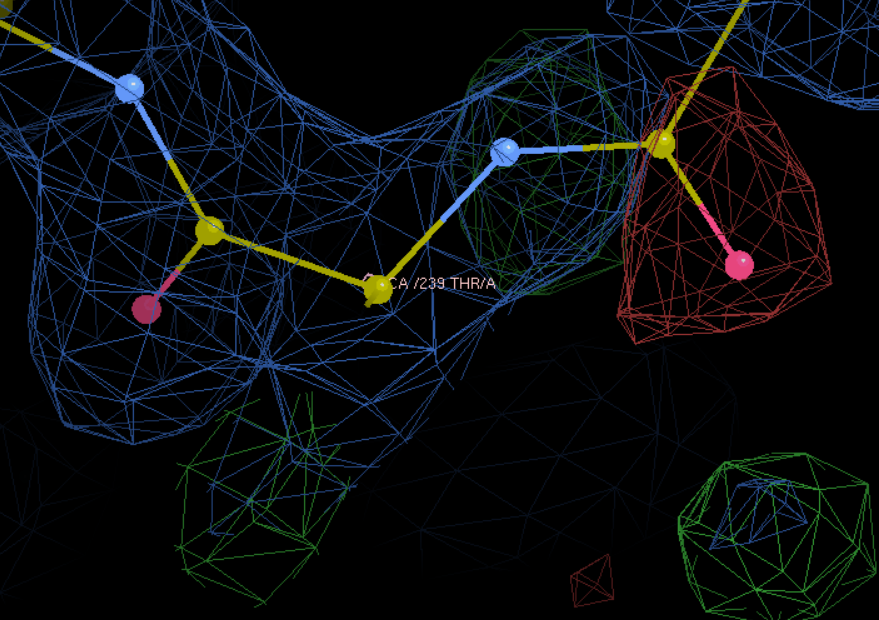
L改为F
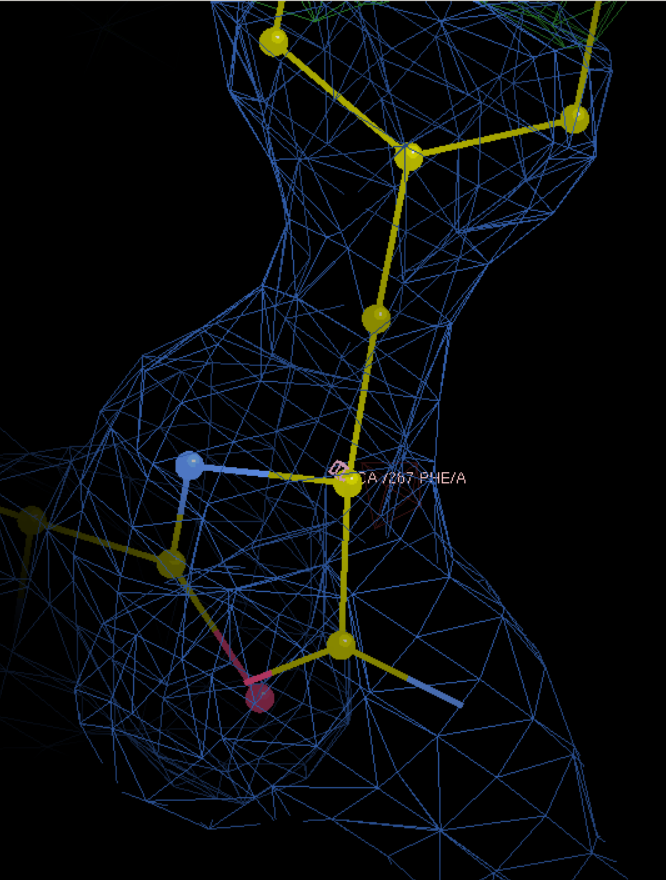
R改为S
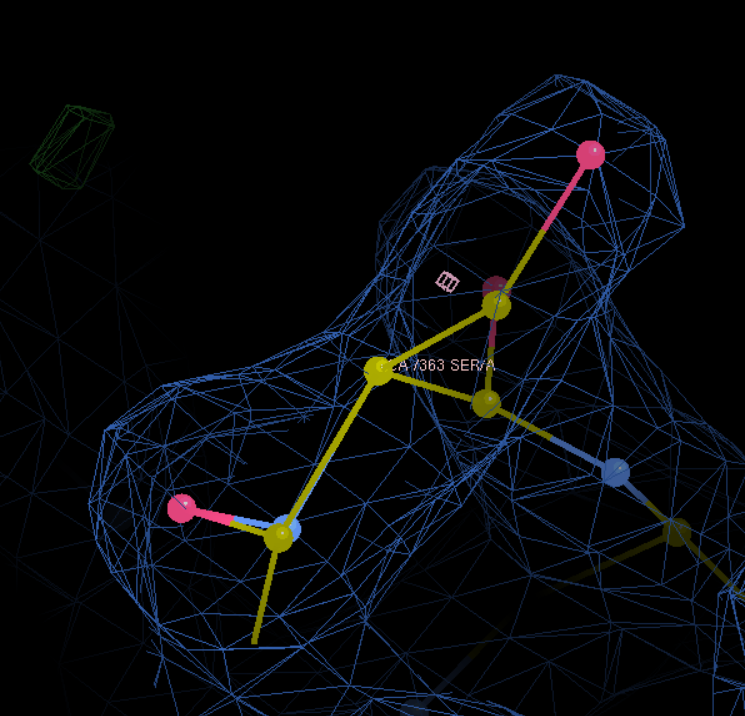
(2)然后就是在活性中心添加HEM
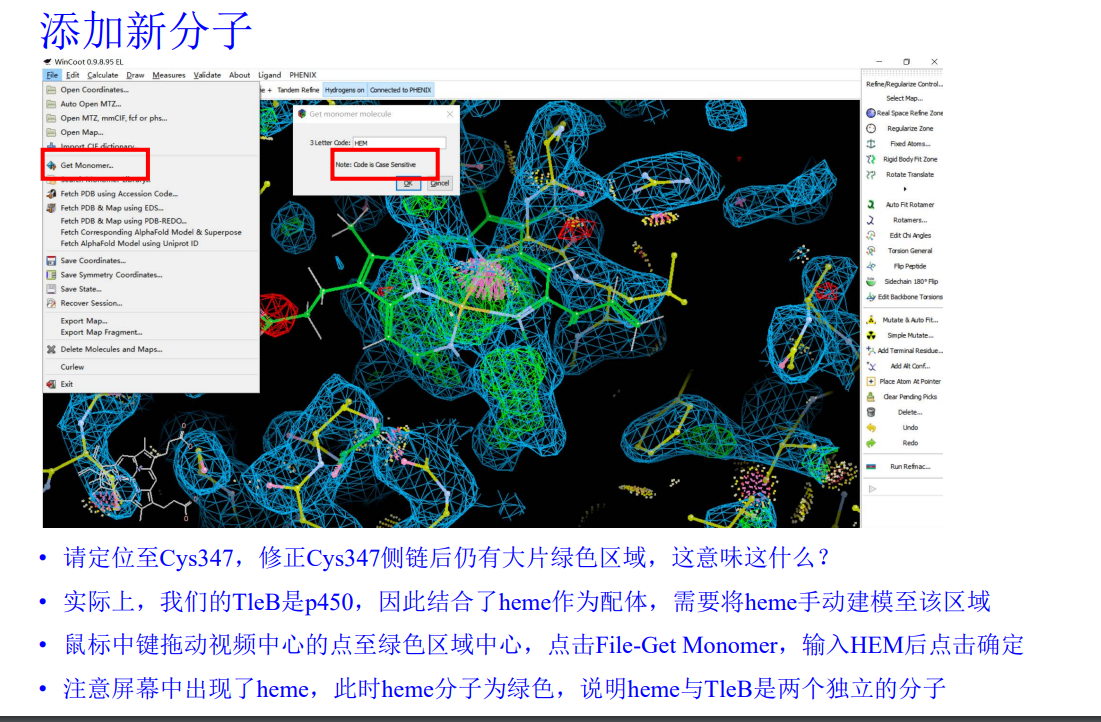
可以看到已经放进去了:
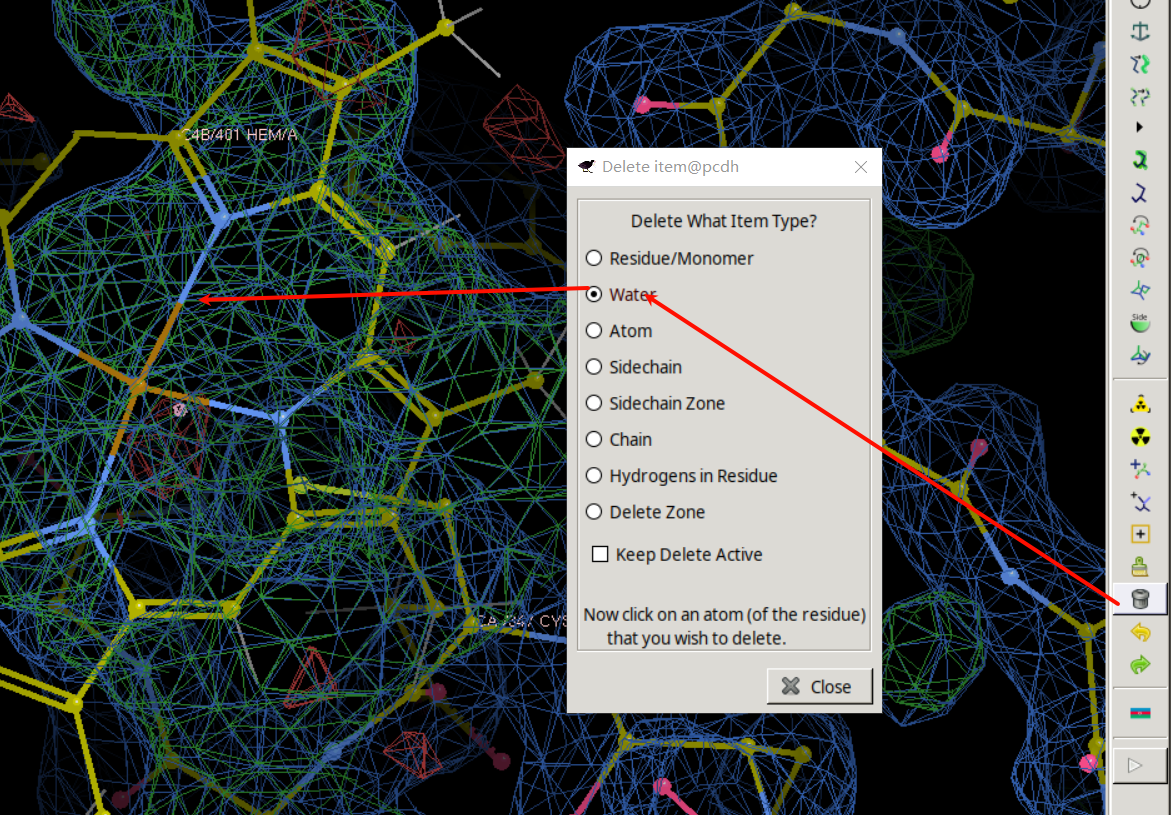
手动进入347 Cys
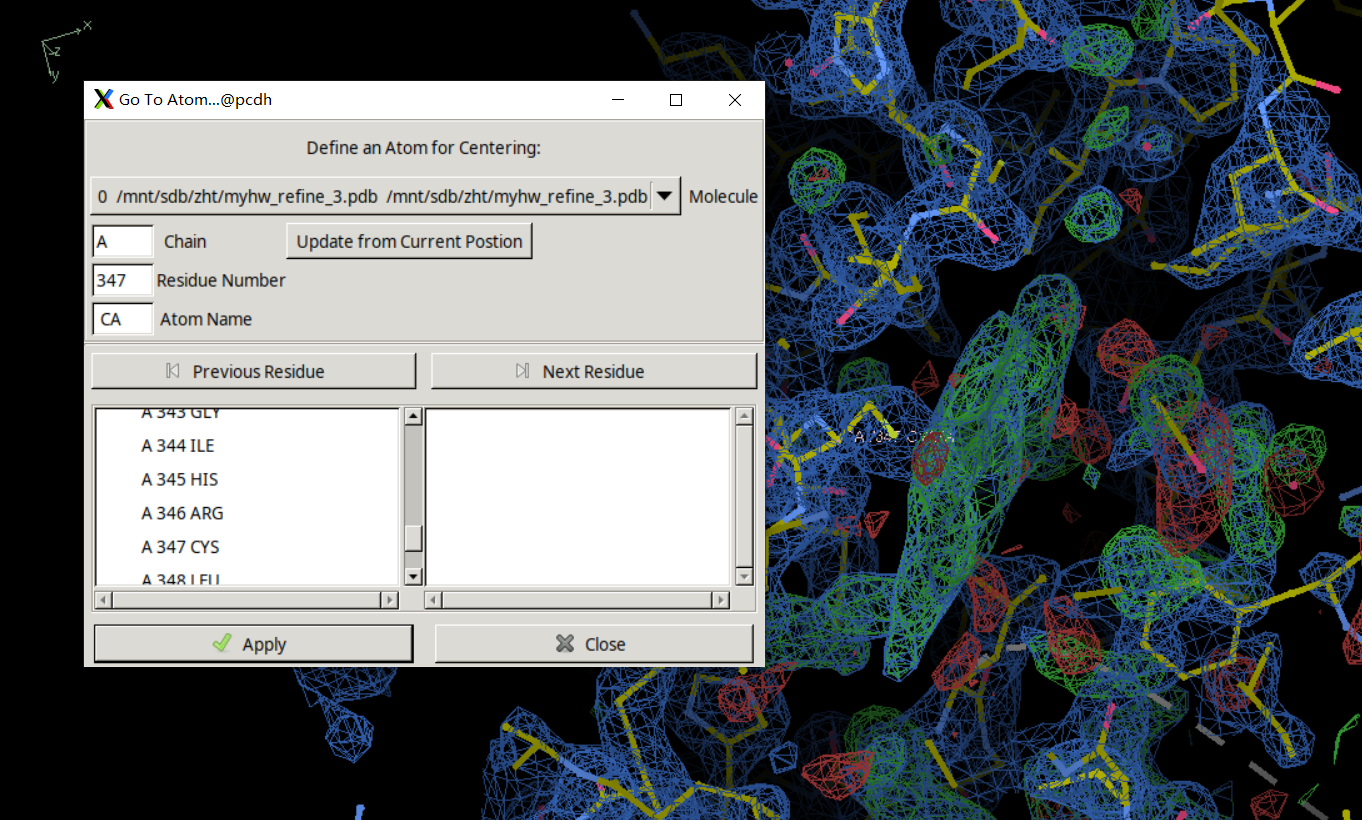
就是这里有一大片绿色的电子云,在这里添加HEM
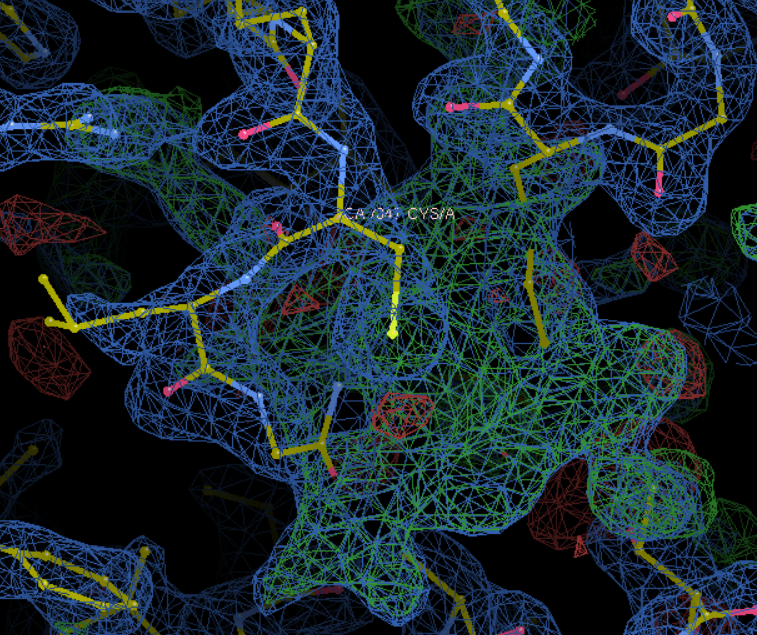
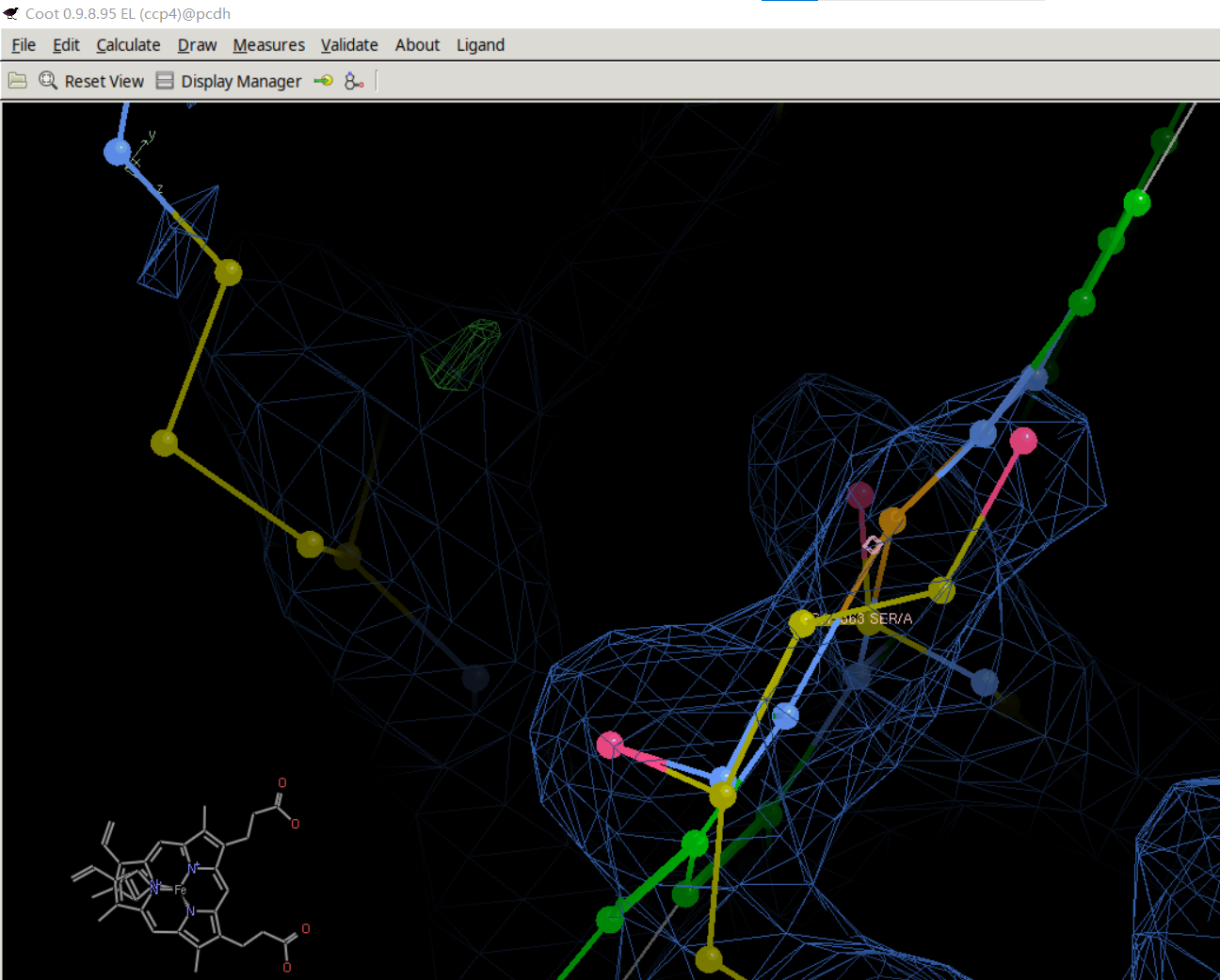
然后我们在这里加入了HEM
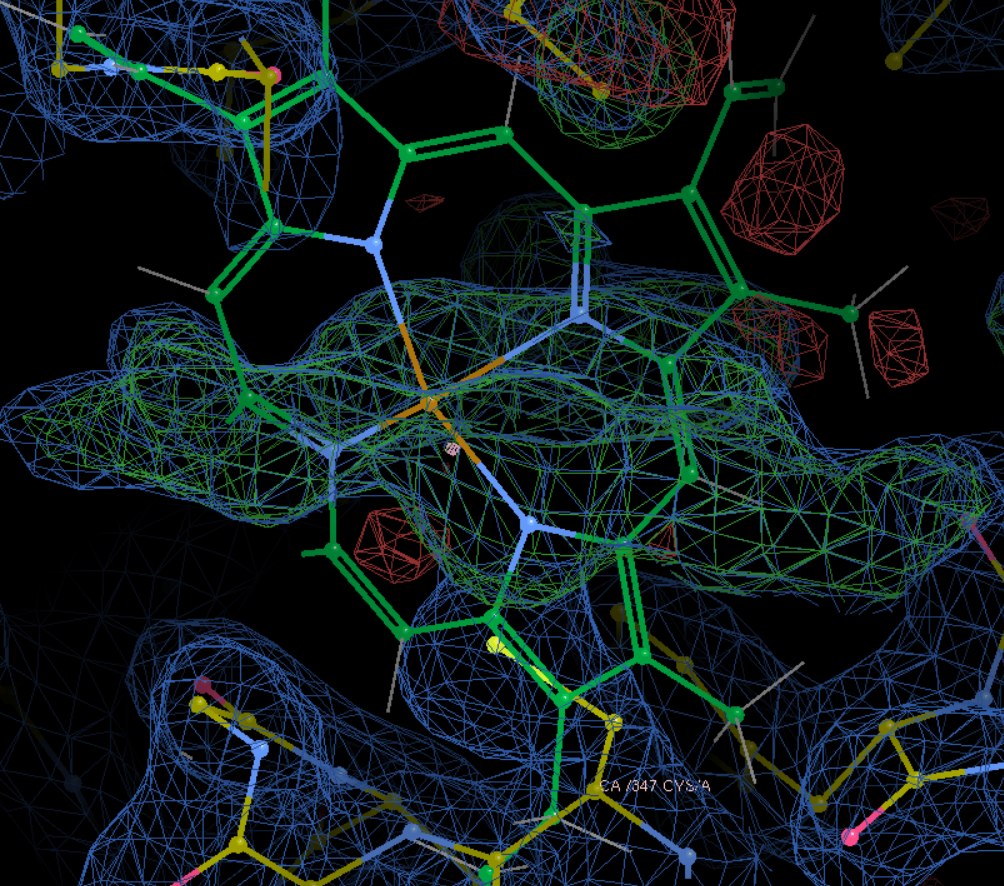
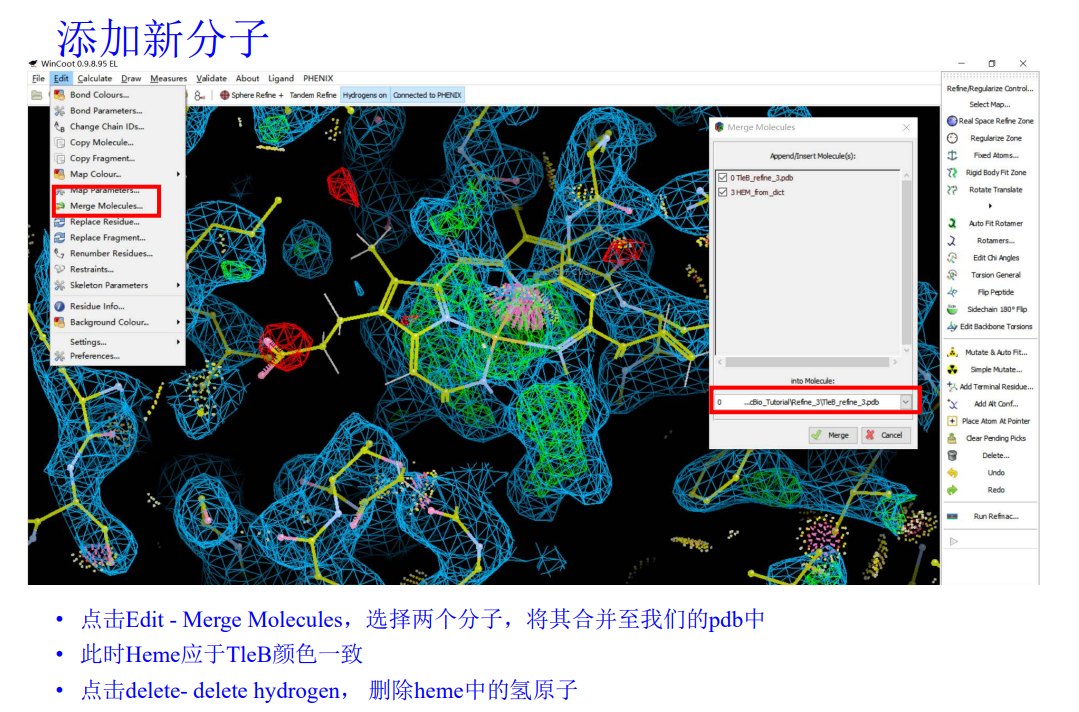
合并之后进行微调:

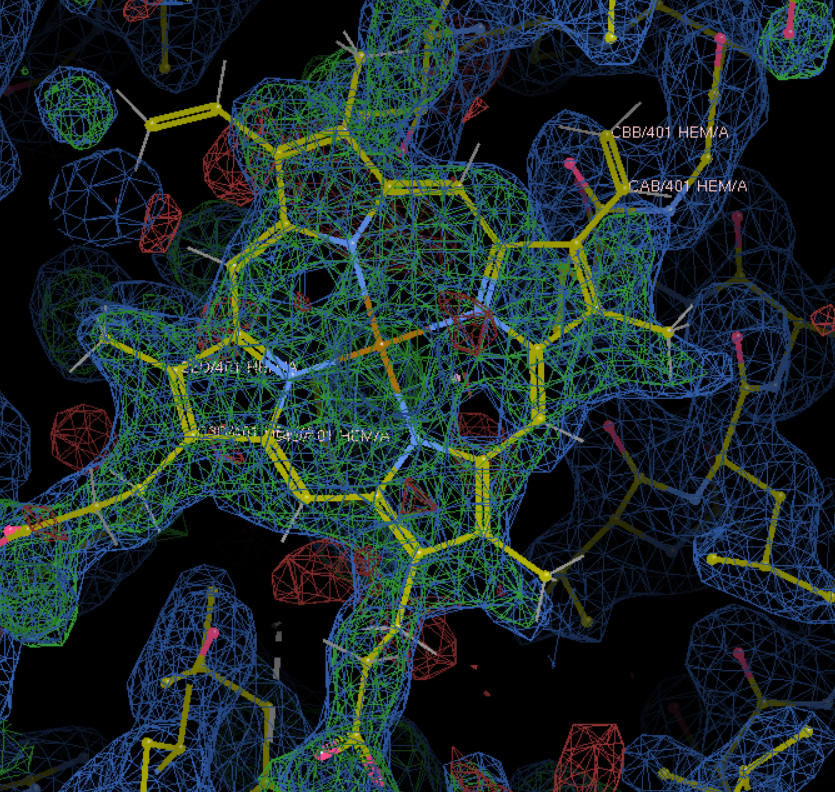
最后初步精修效果大致如下:
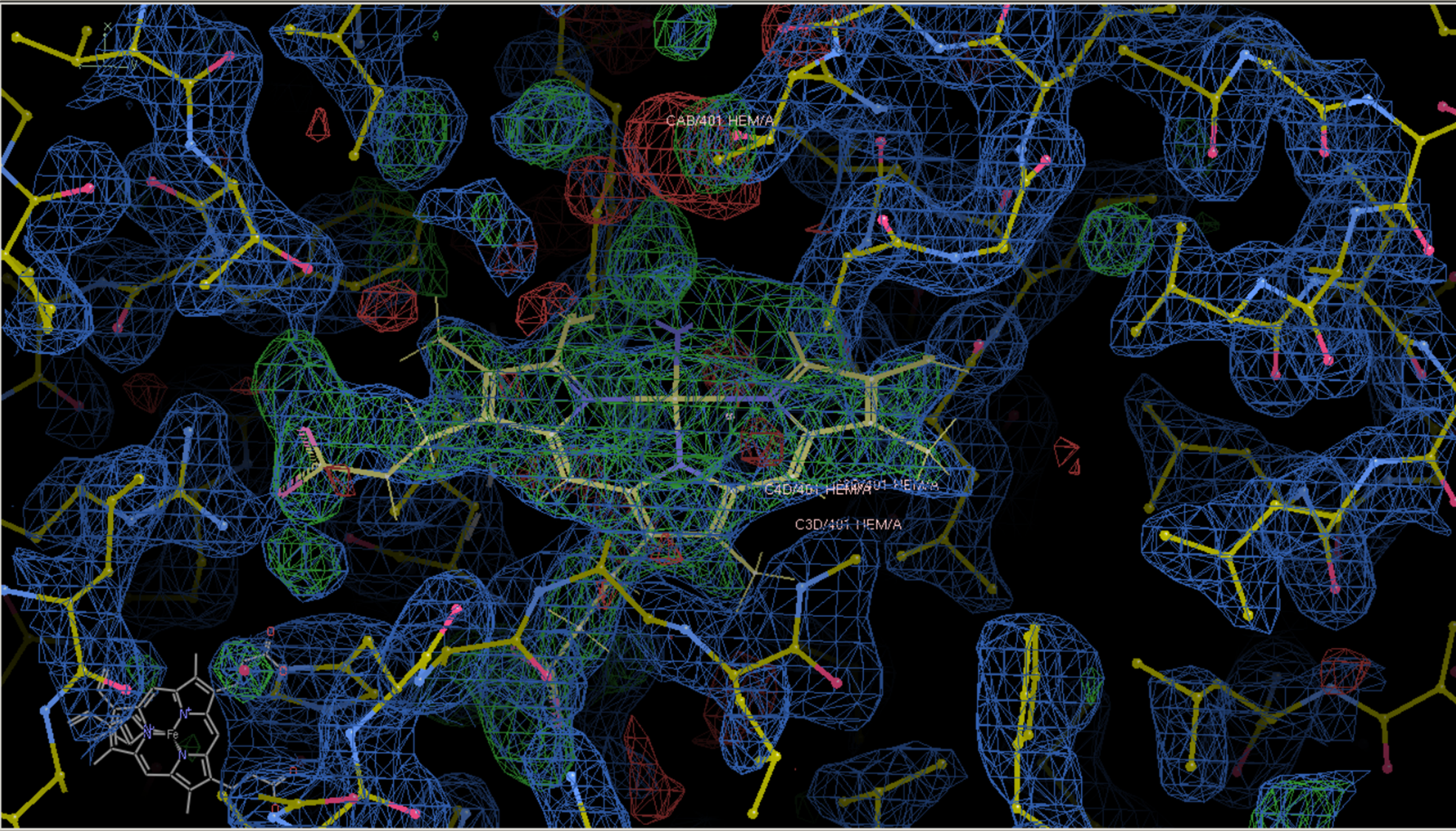
HEM这个环有些细节还是没有移动上,然后就是随时保存精修之后的模型文件,也就是pdb文件
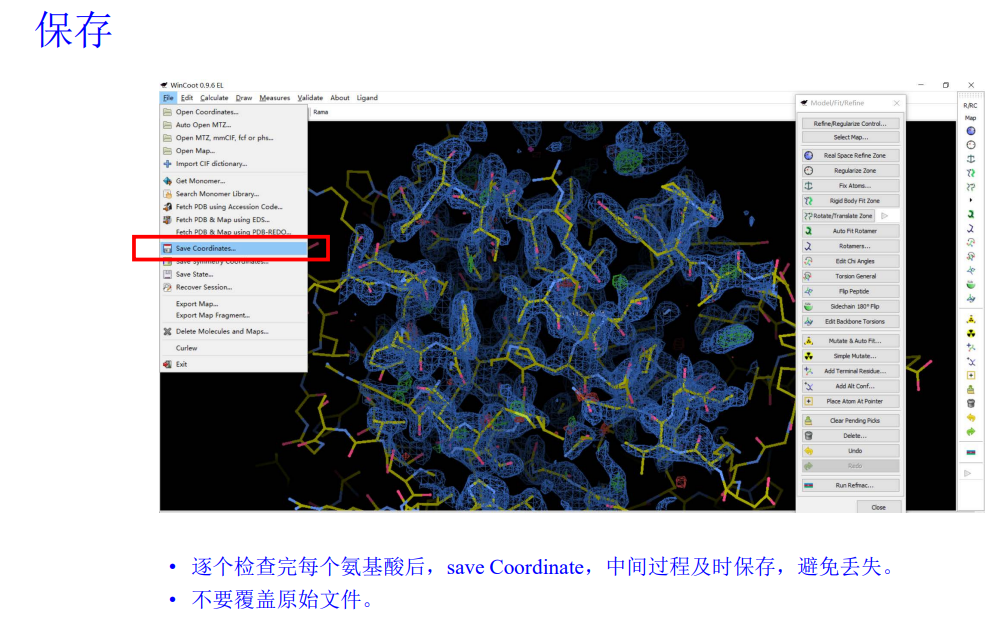
7,然后再用phenix的refine进行机器精修:
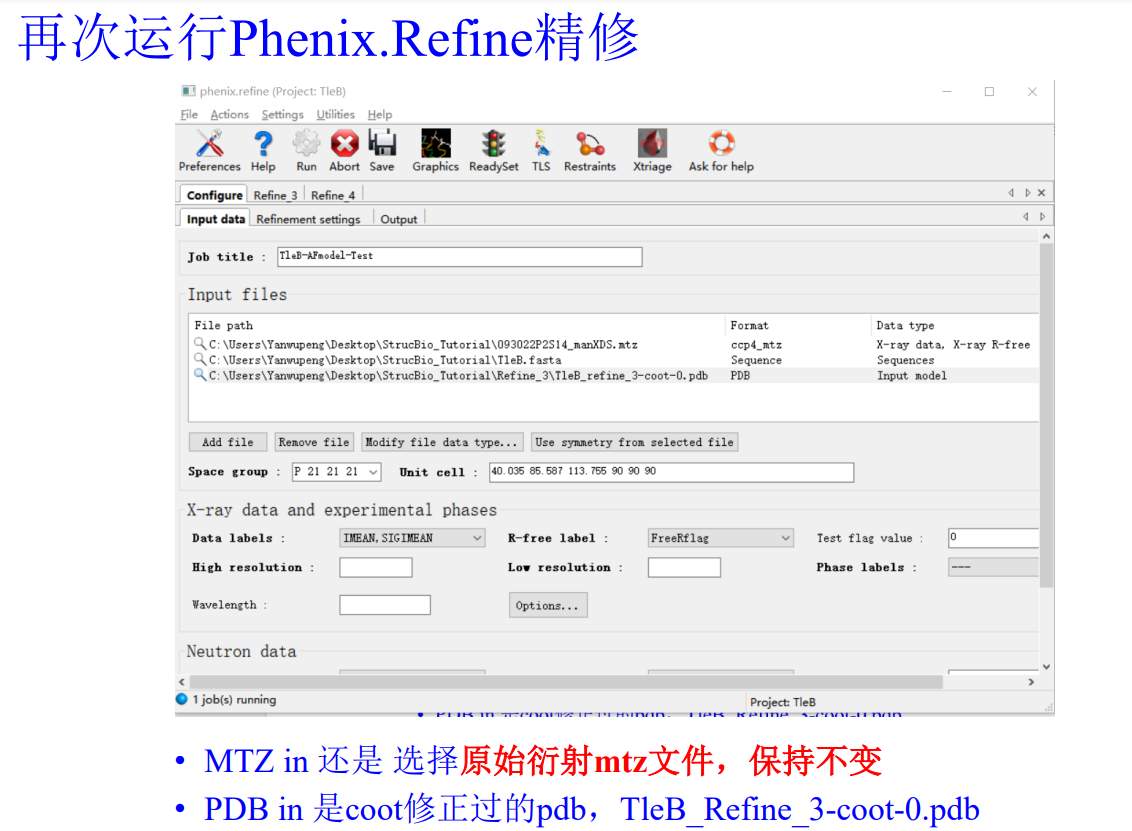
还是打开job history:然后还是refine这个界面,在input files上修改一下,主要是pdb的结构模型文件要使用此次精修之后的文件
或者在home中打开如下:
然后就是run,也就是下一轮机器的精修
精修结果:
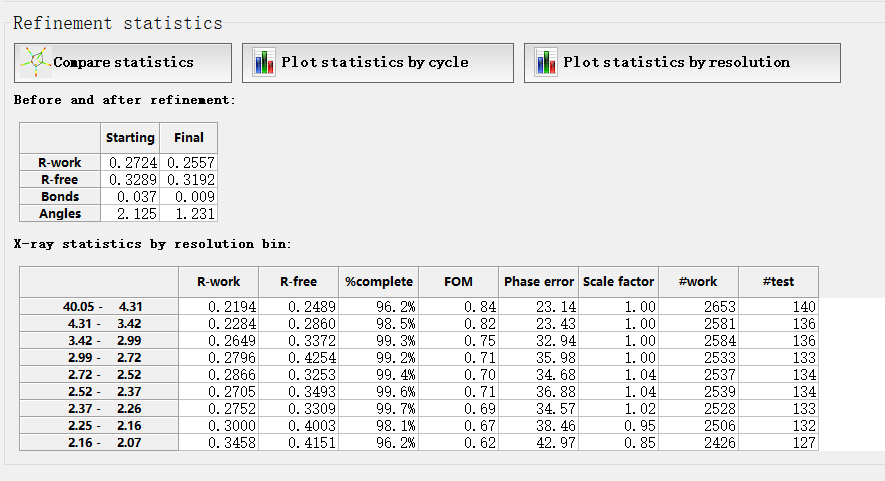
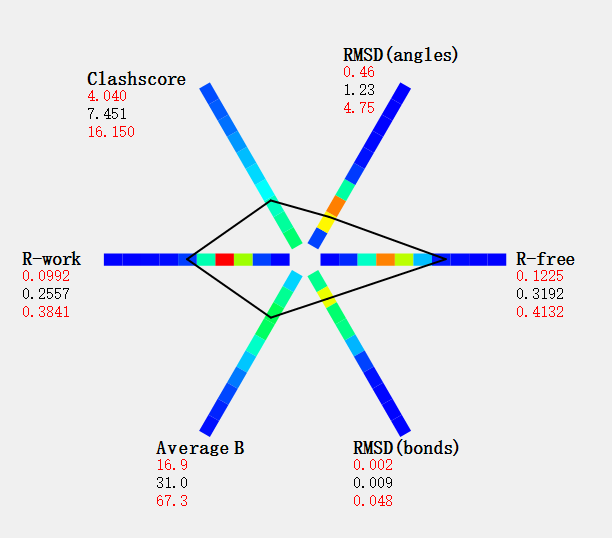
其他的结果:
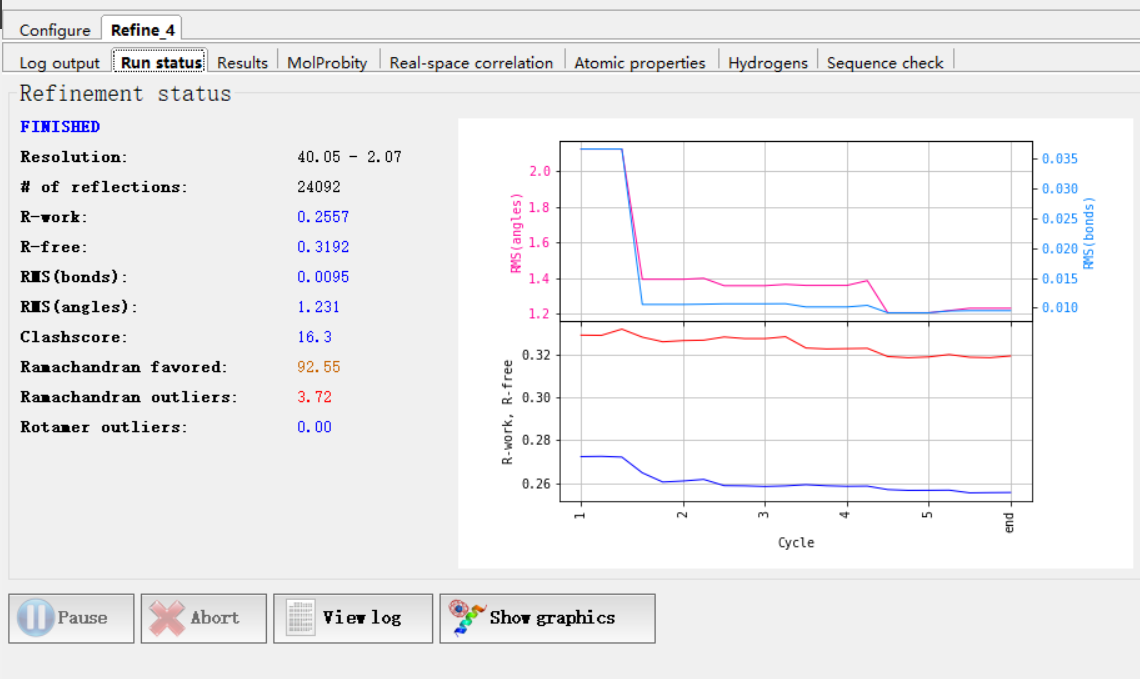

等
8,validation:
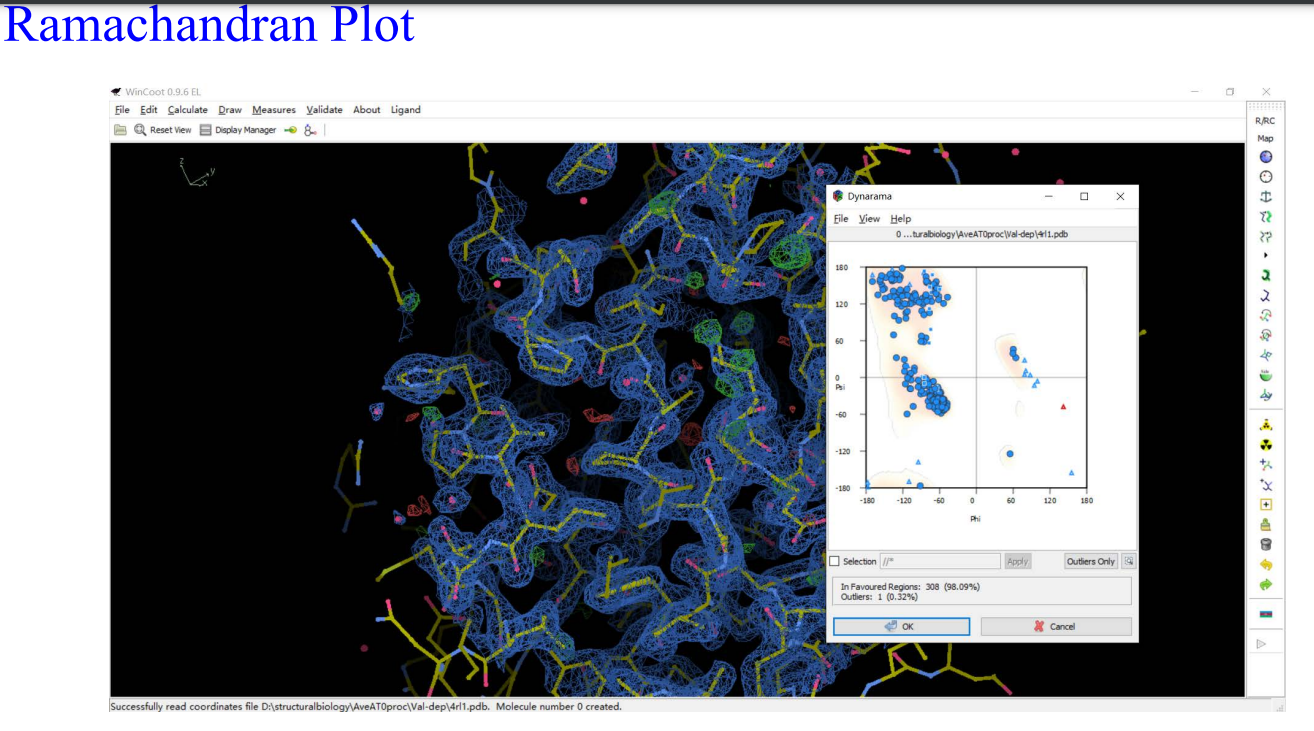
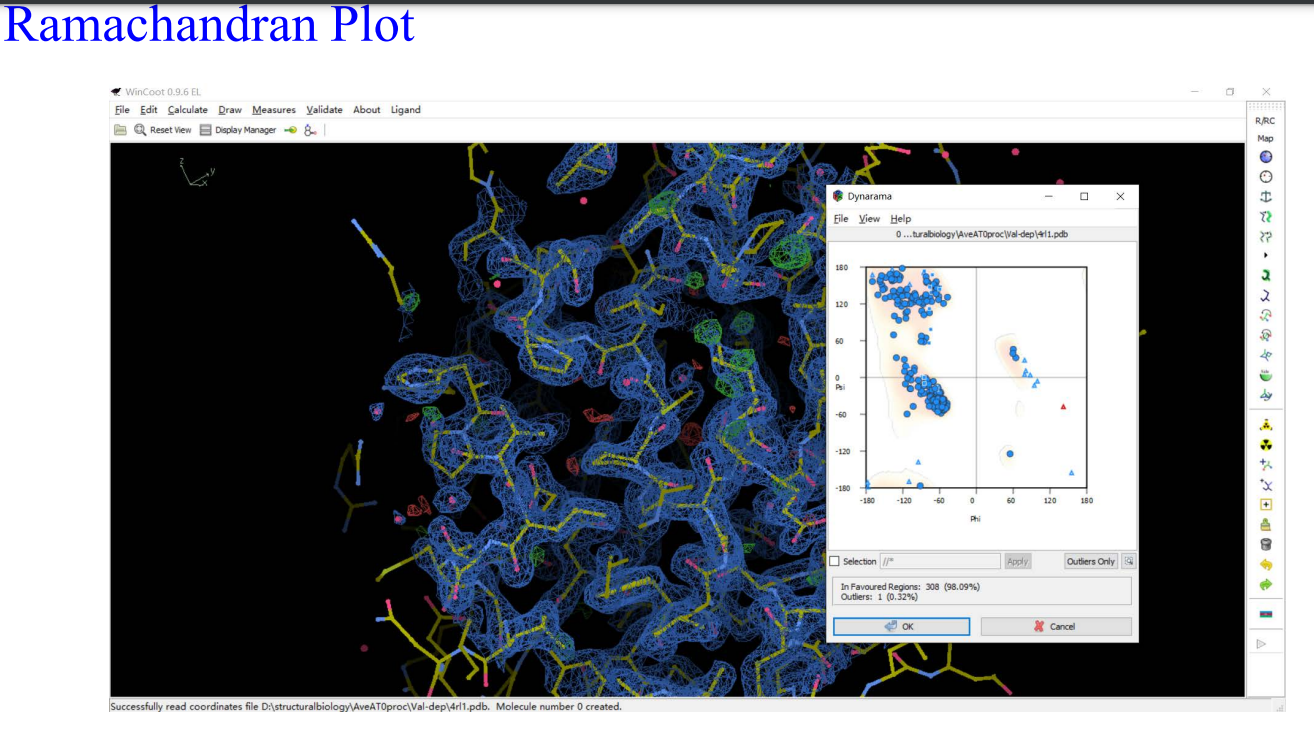
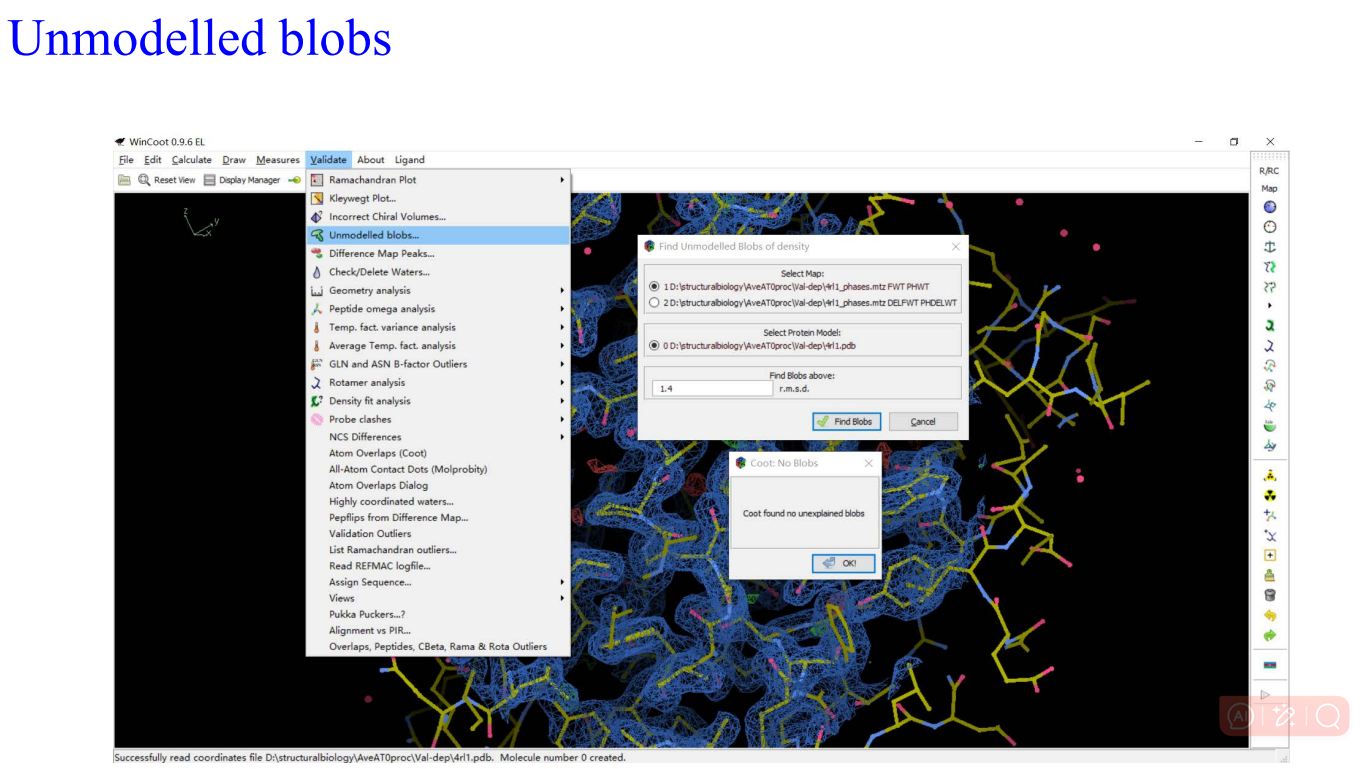
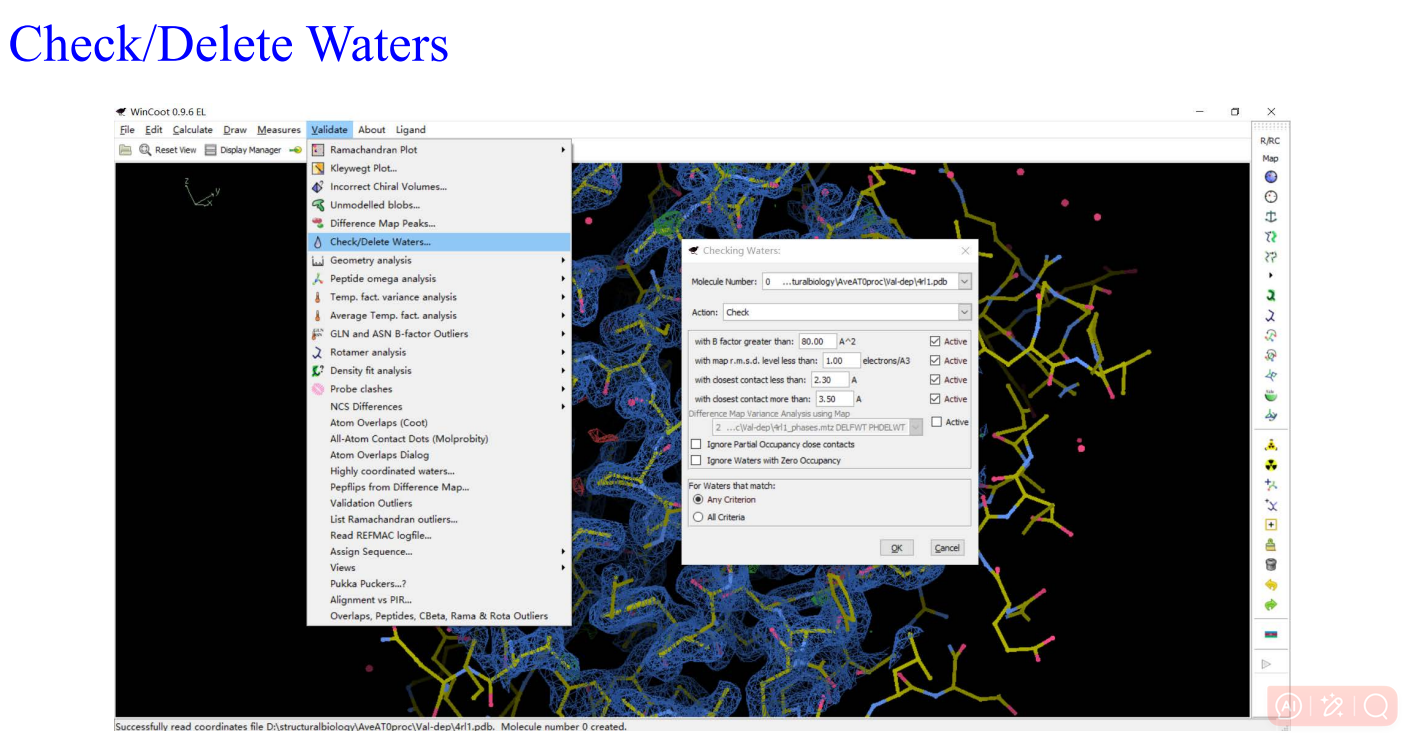
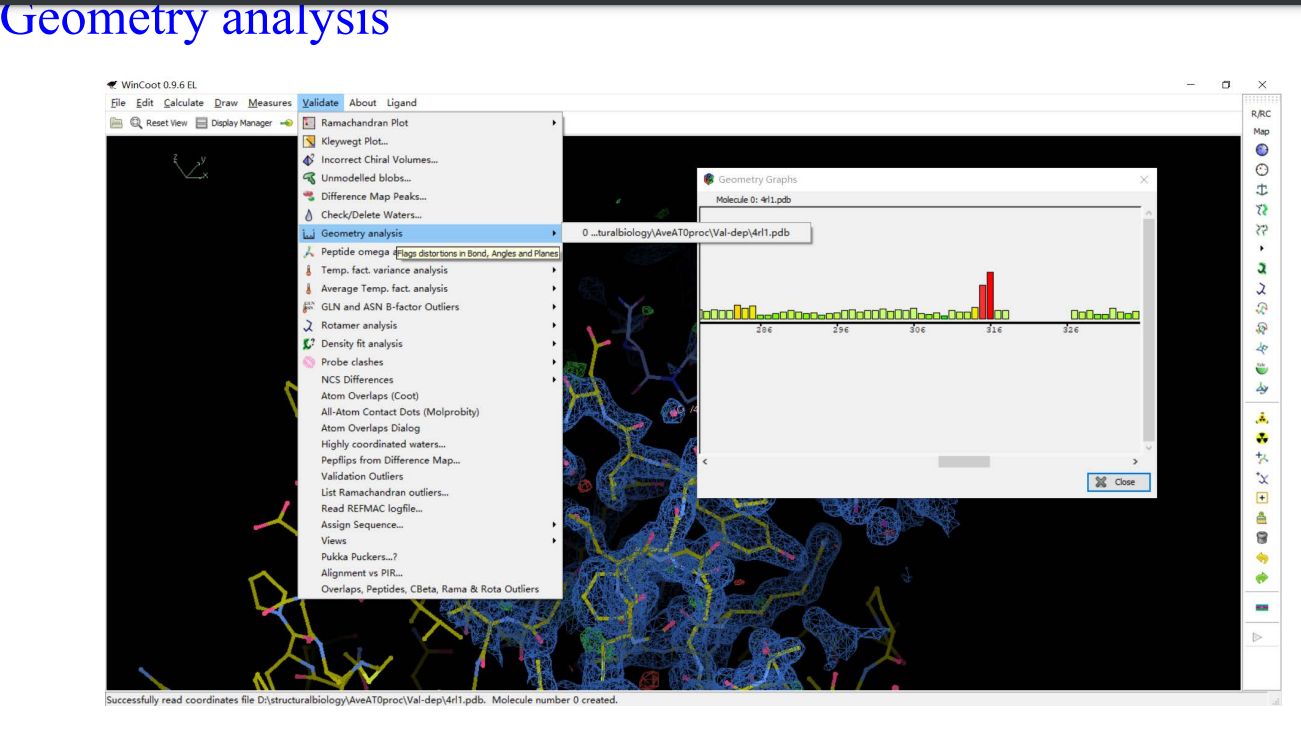
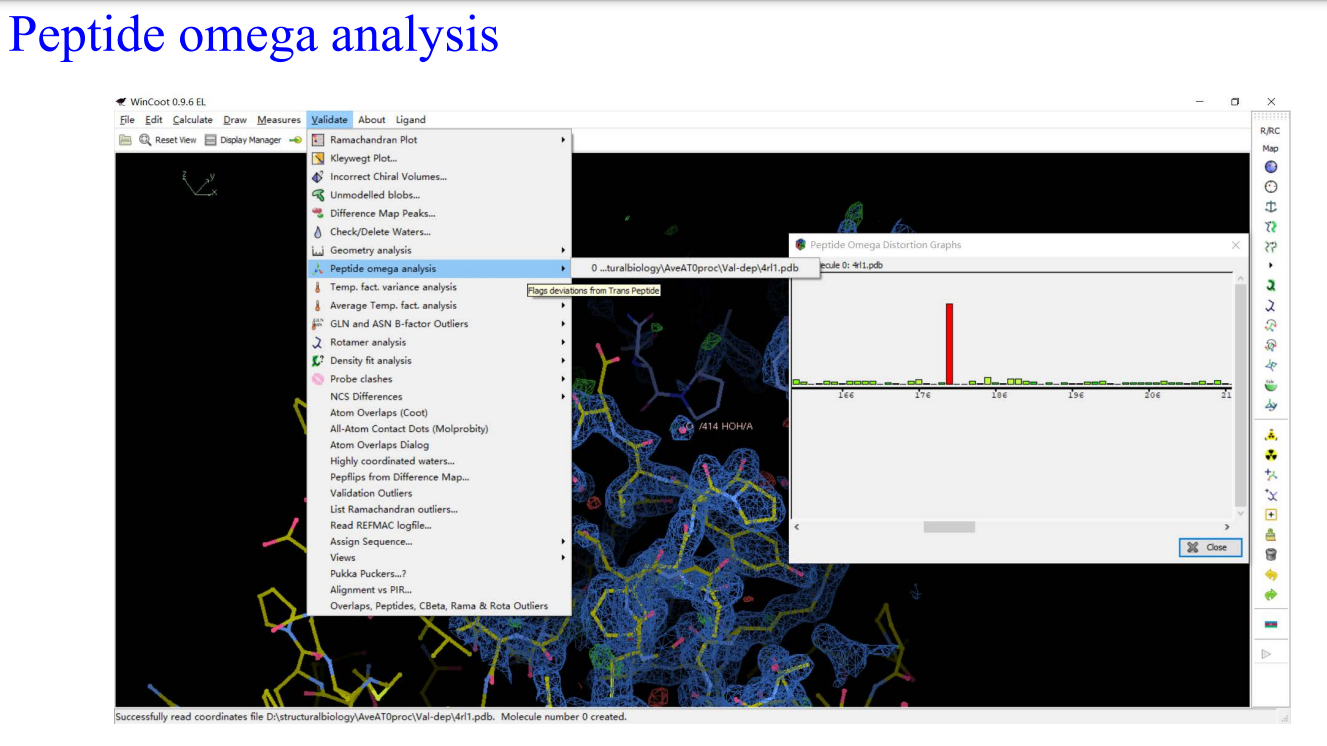
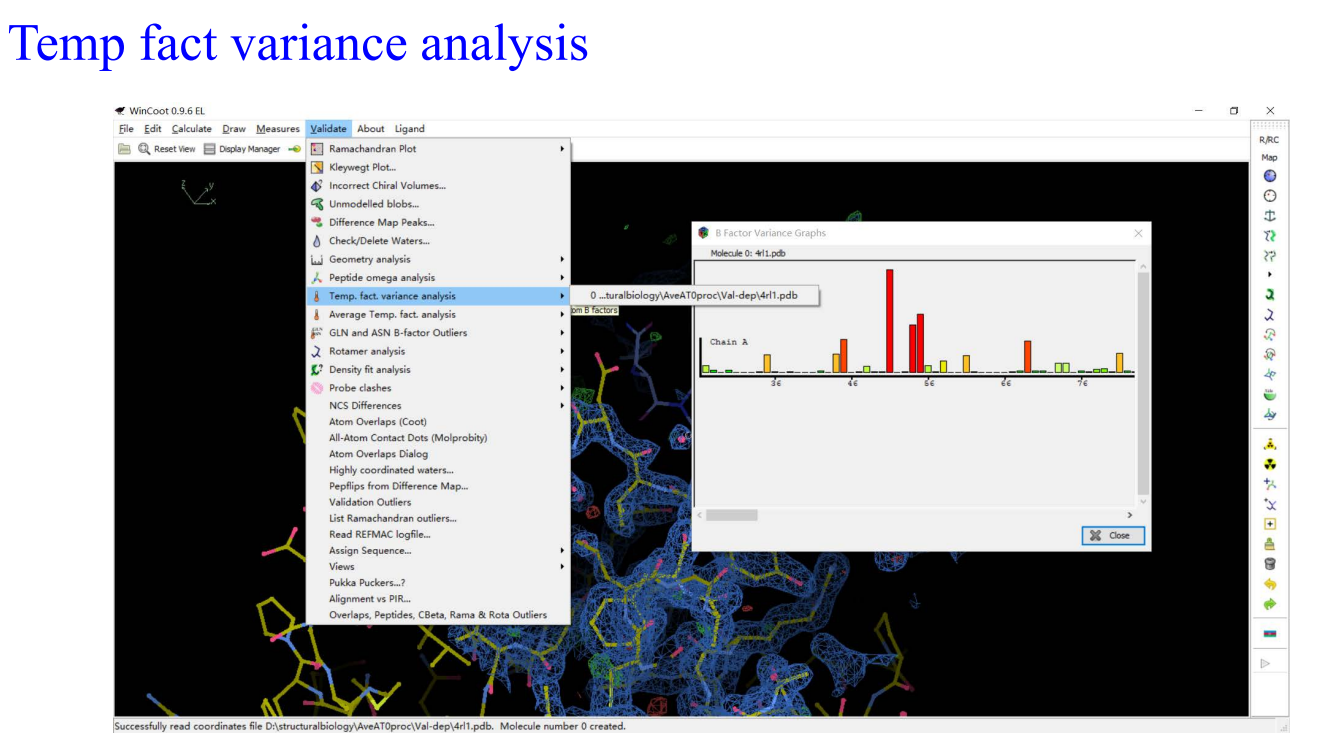
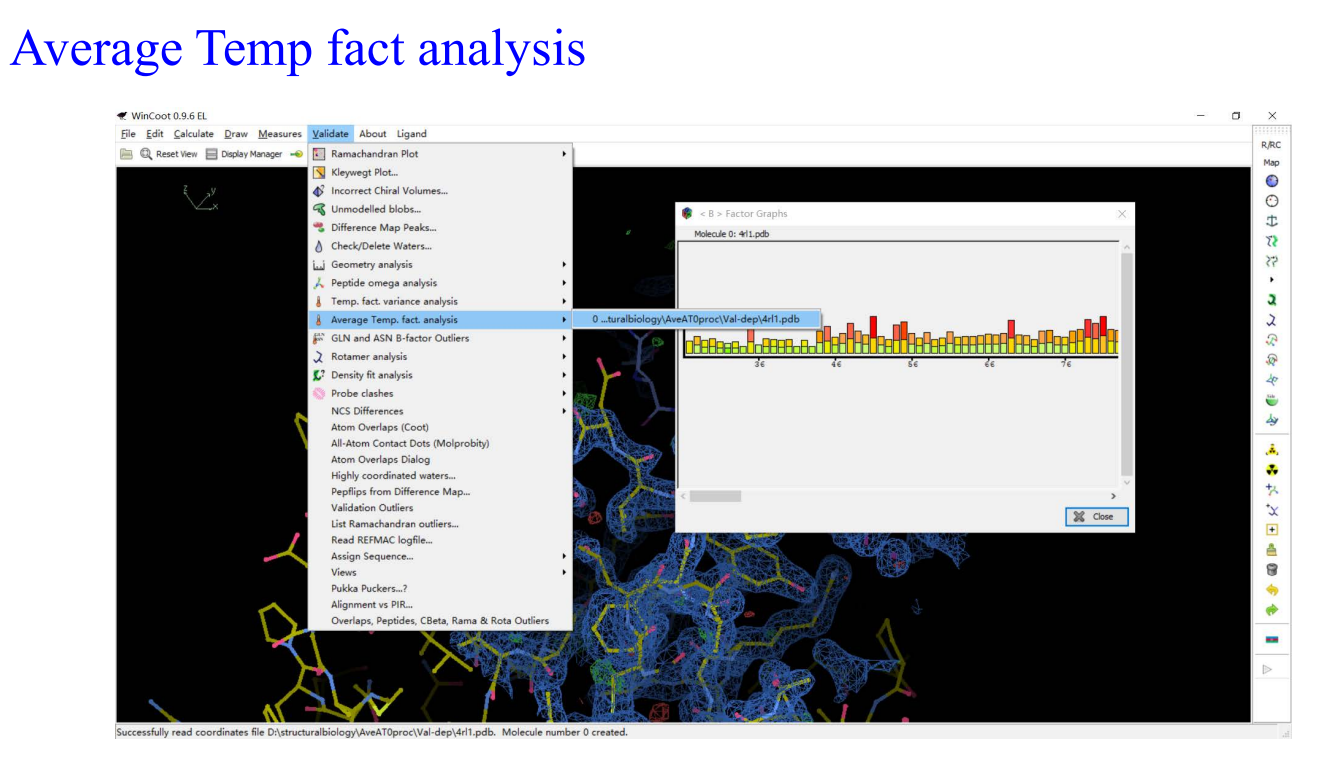
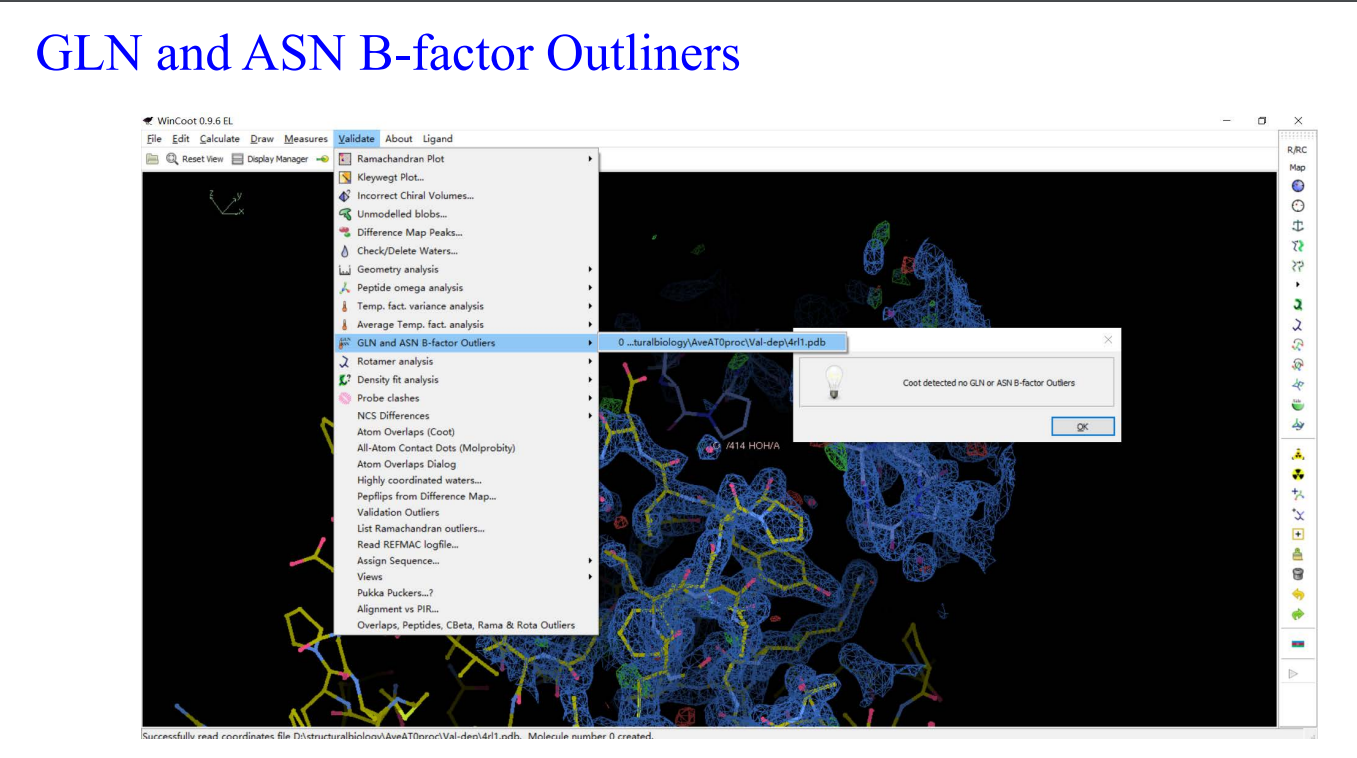
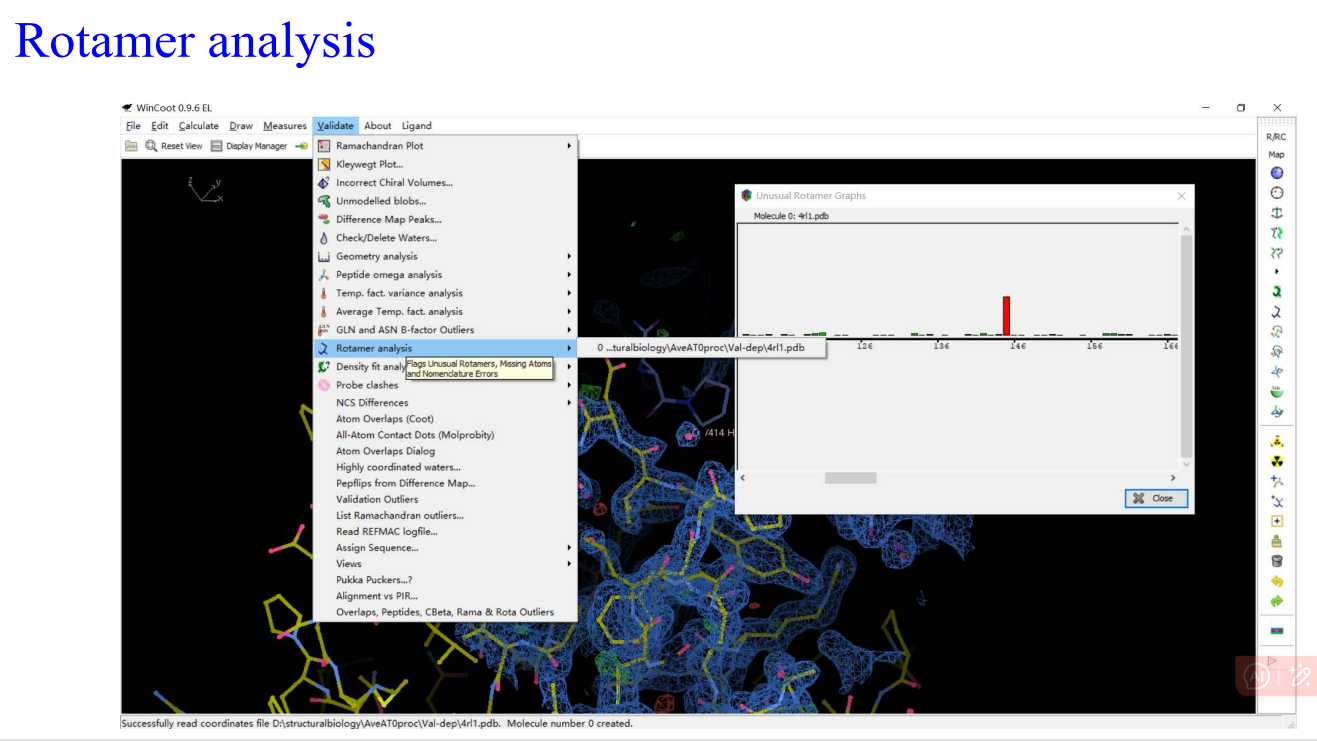
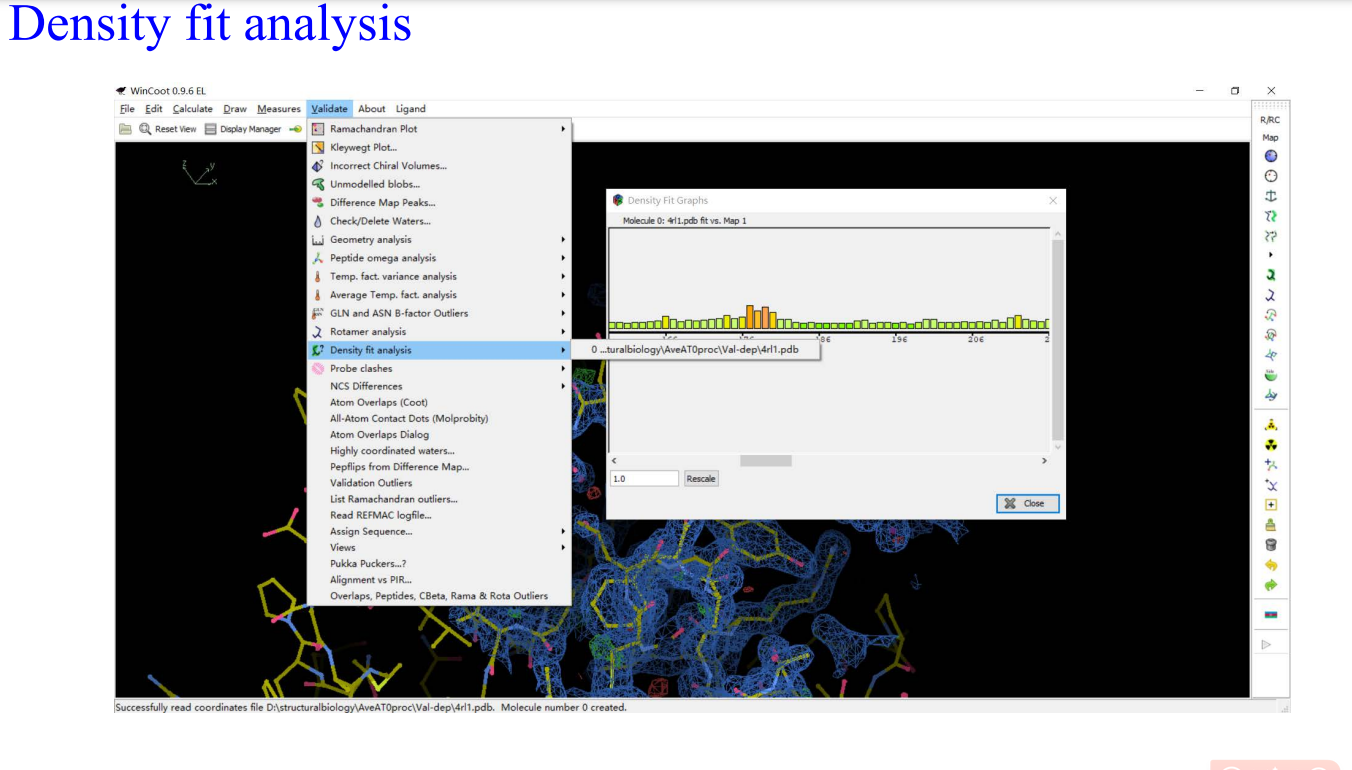
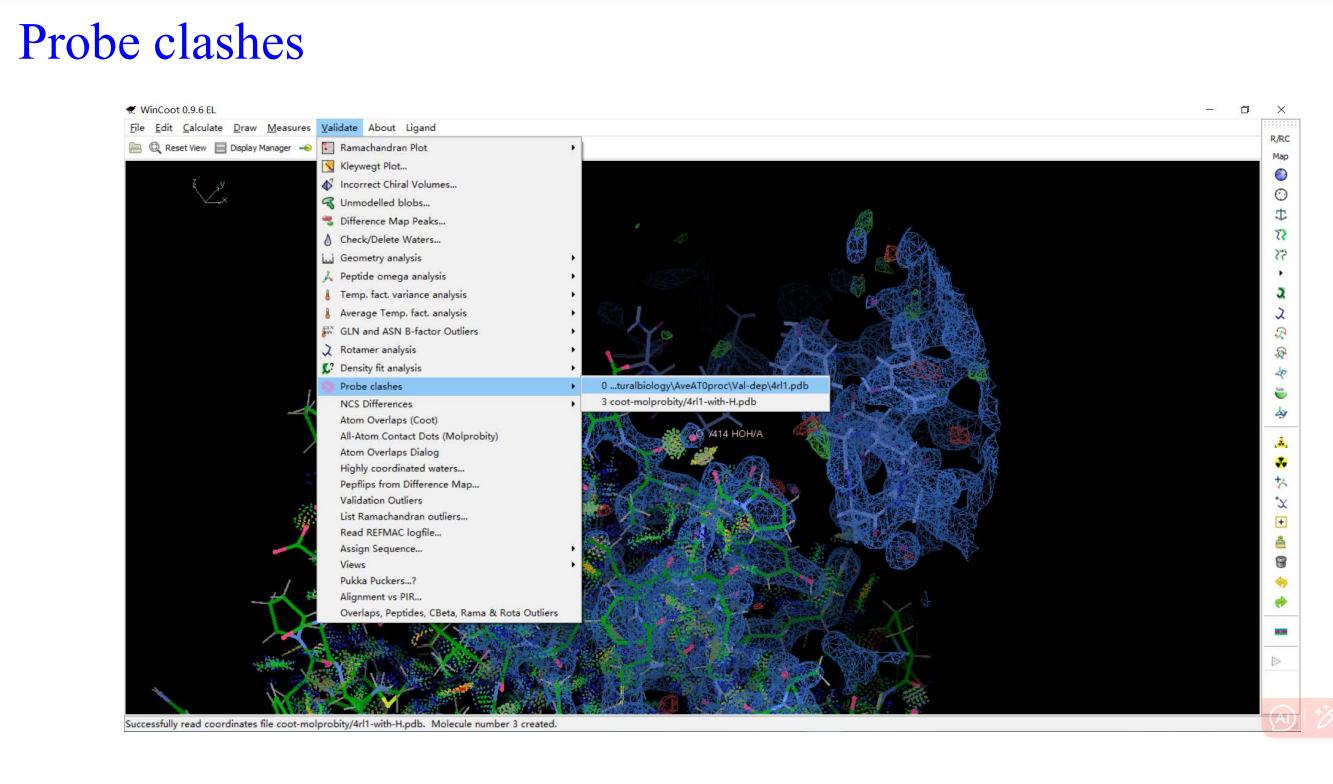
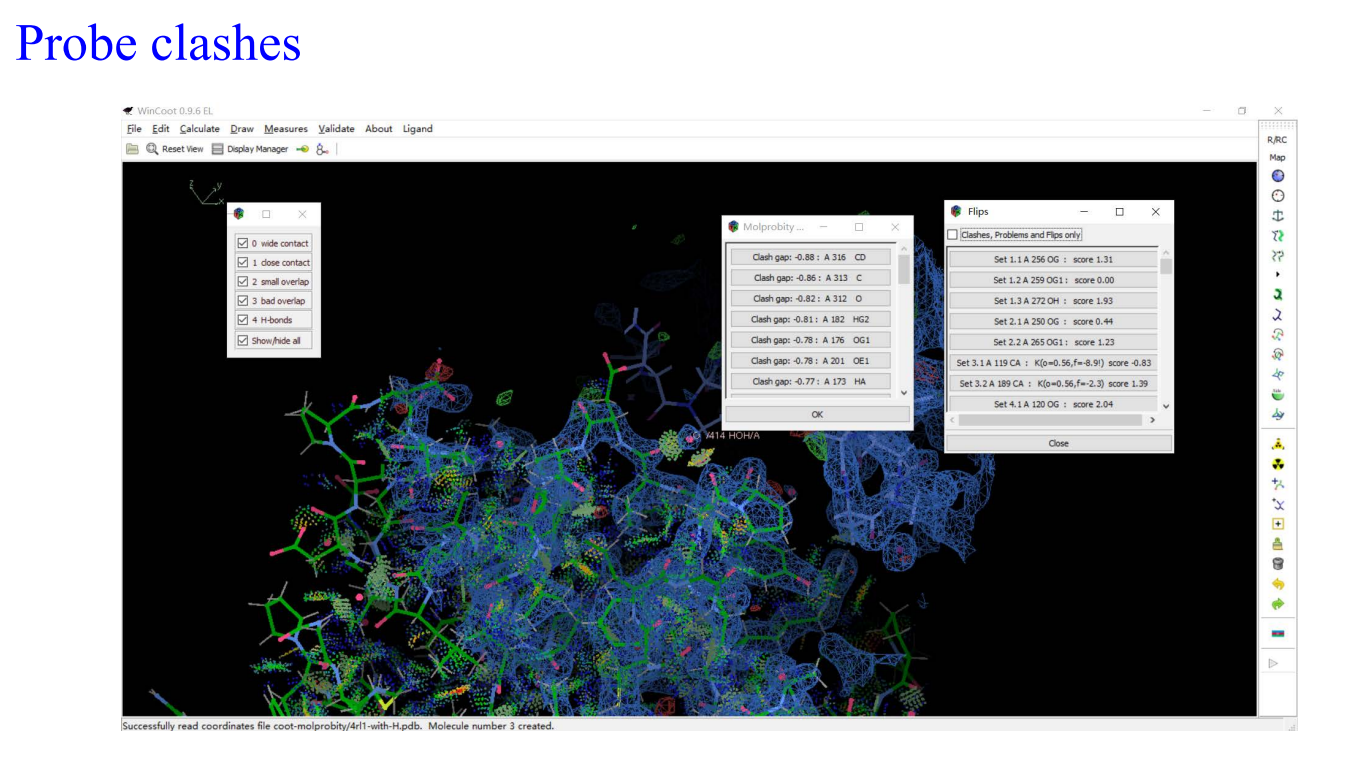
9,Molprobity:
其实这个模块在phenix中也是内置的,但是还是推荐在phenix外在线做
http://molprobity.biochem.duke.edu/index.php
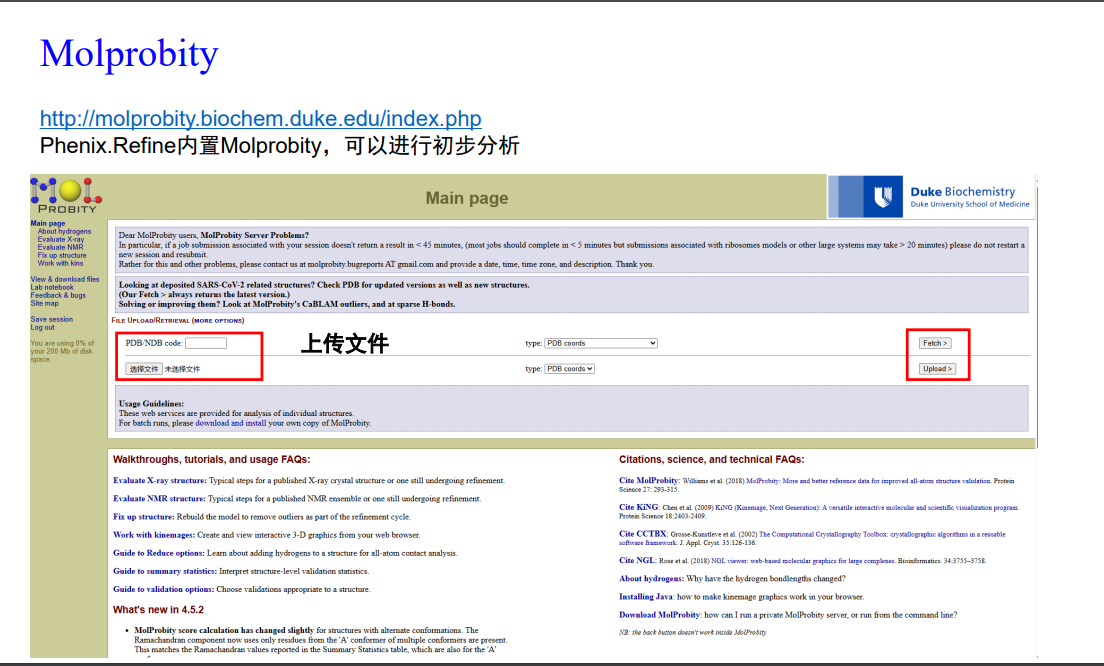
然后注意上一步精修之后的结果放在文件夹:使用refine_4的pdb模型文件
选择上面精修好的模型pdb文件(实际过程中可以精修好多轮,假设上面的refine已经是我们精修之后达到要求了)
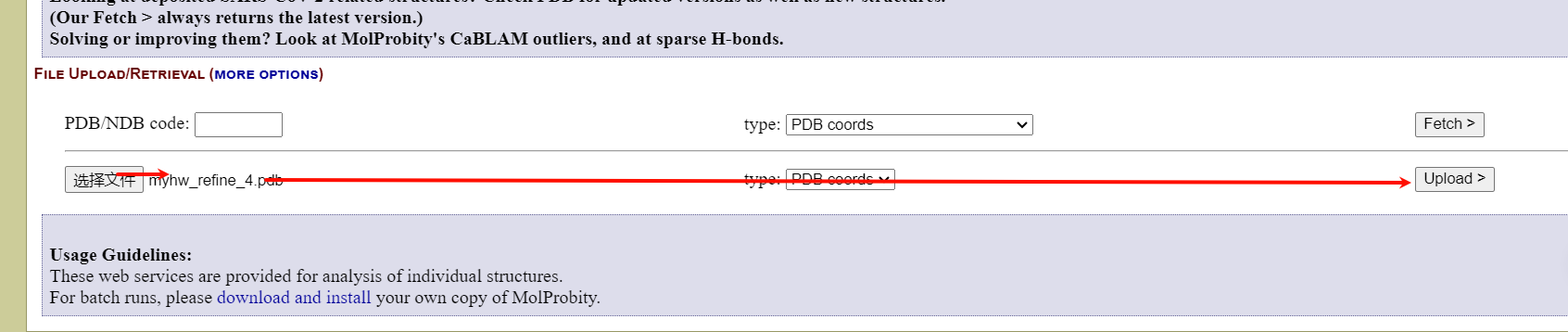
如果页面没反应就再上传pdb文件,直到跳转到界面:


点击continue:
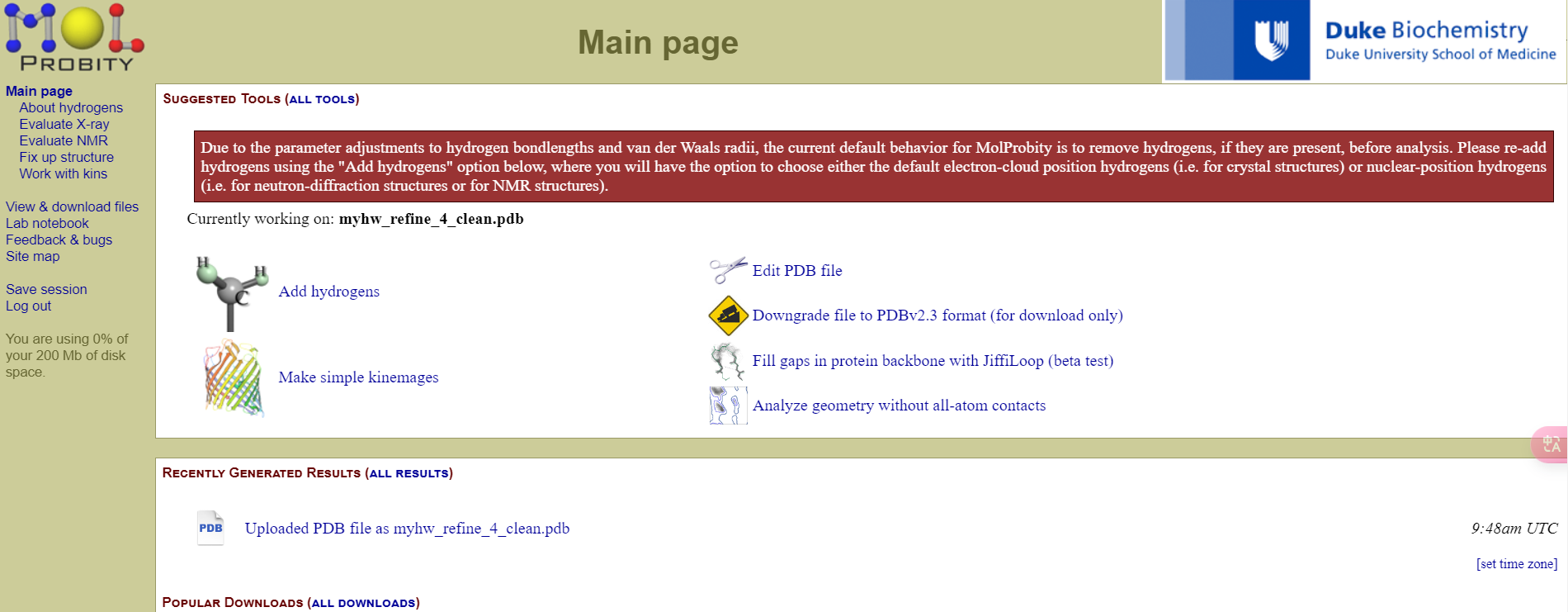
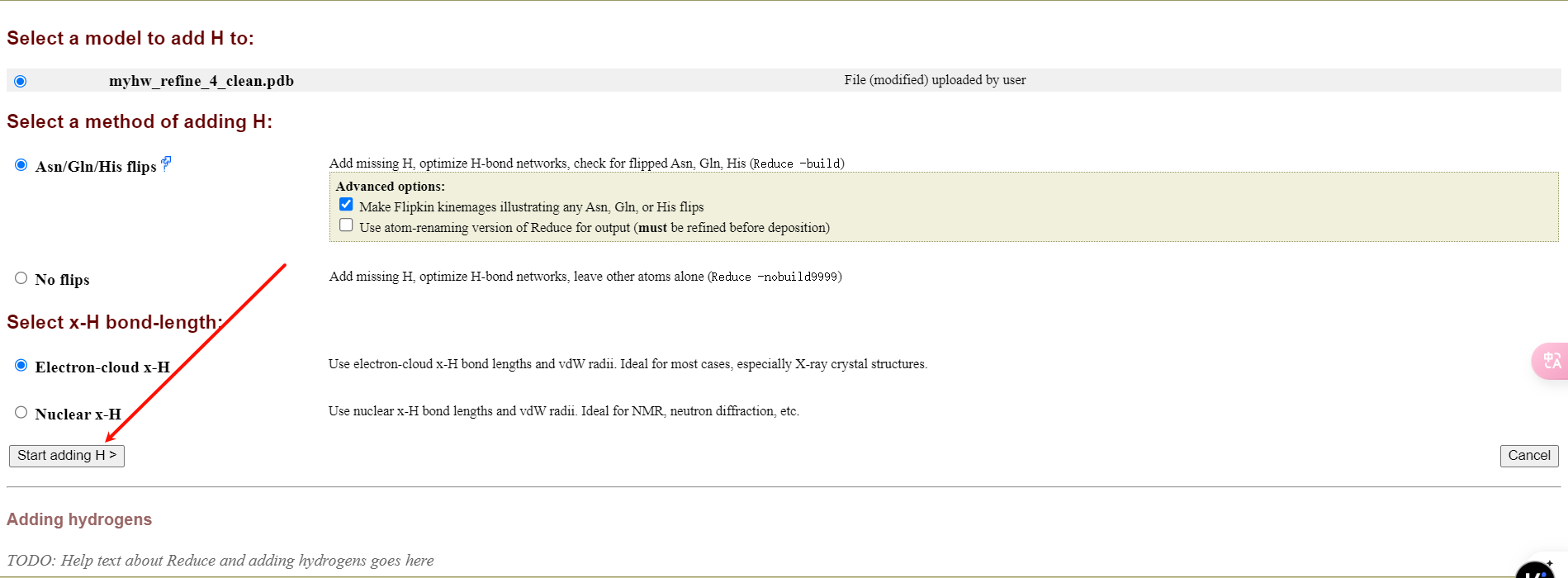
首先是加氢add hydrogens
一般默认即可
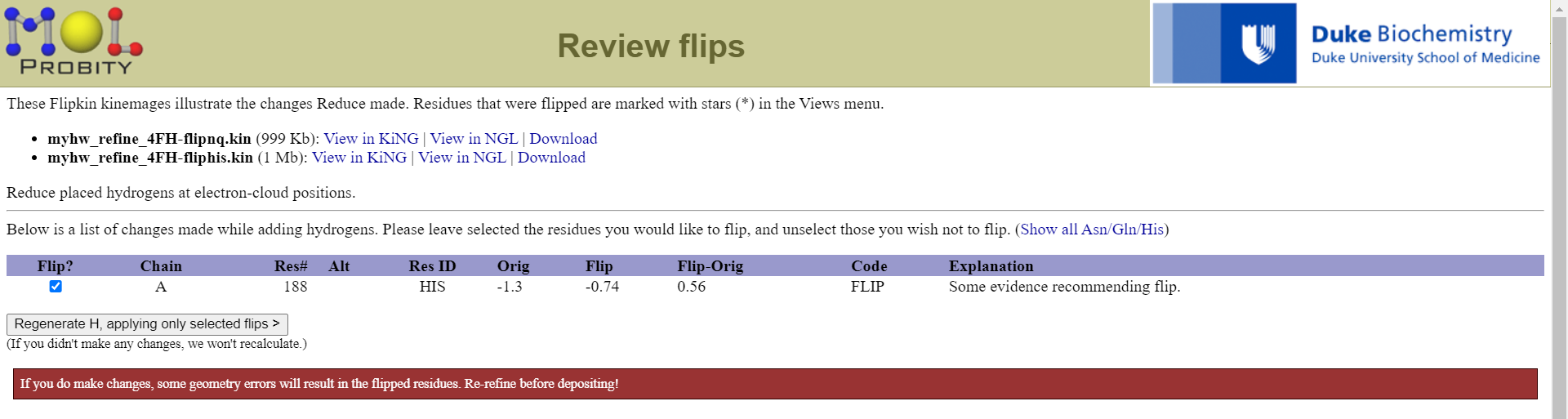
然后机器判断我们这里188号位的His氨基酸侧链需要翻转,我们可以直接接受,然后它会帮助我们生成一个修改之后的pdb文件
点击continue
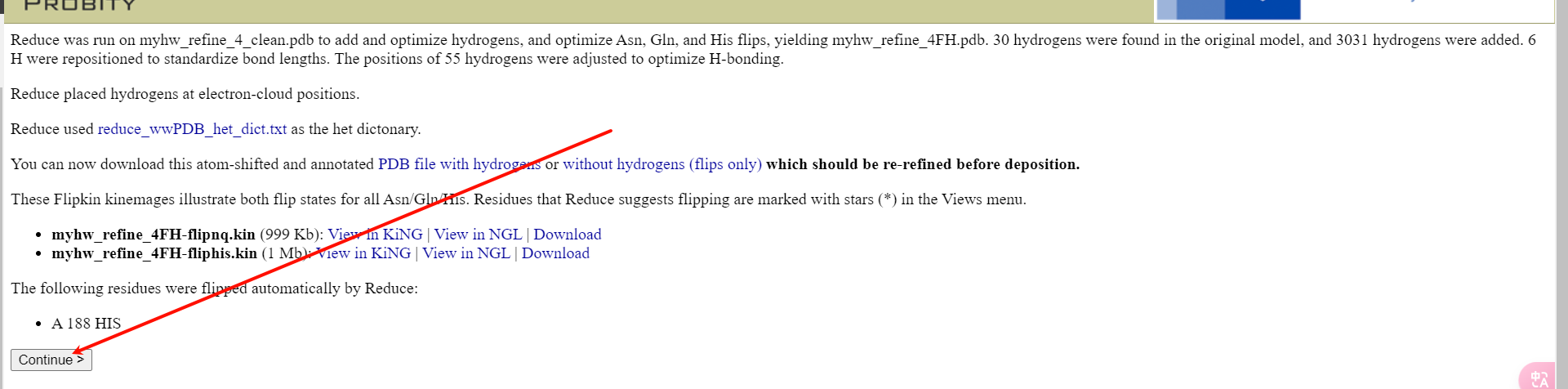
然后选择左上角的这个即可:
然后一切选择默认,直接运行即可
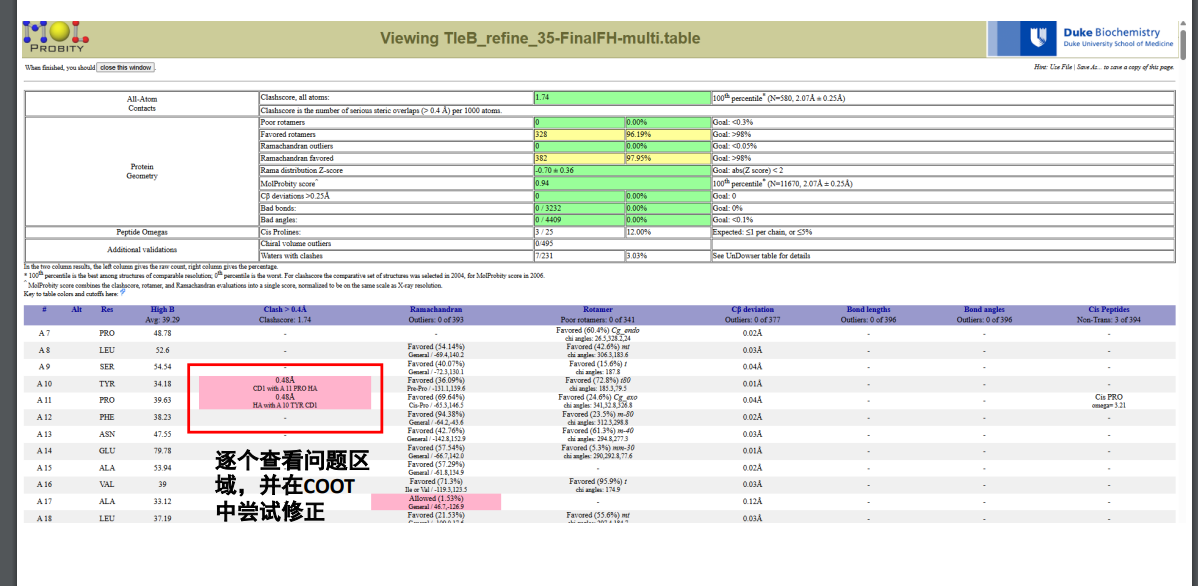
精修结果如下:
绿色代表没有问题,黄色有点问题,红色问题很大
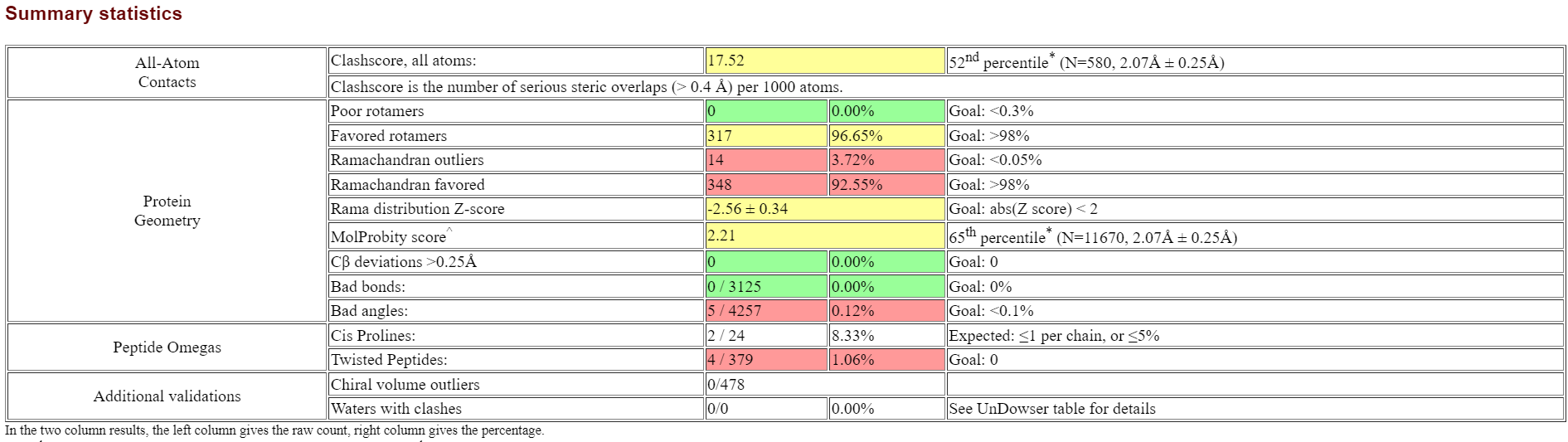
细节部分可以查看下面的这个:
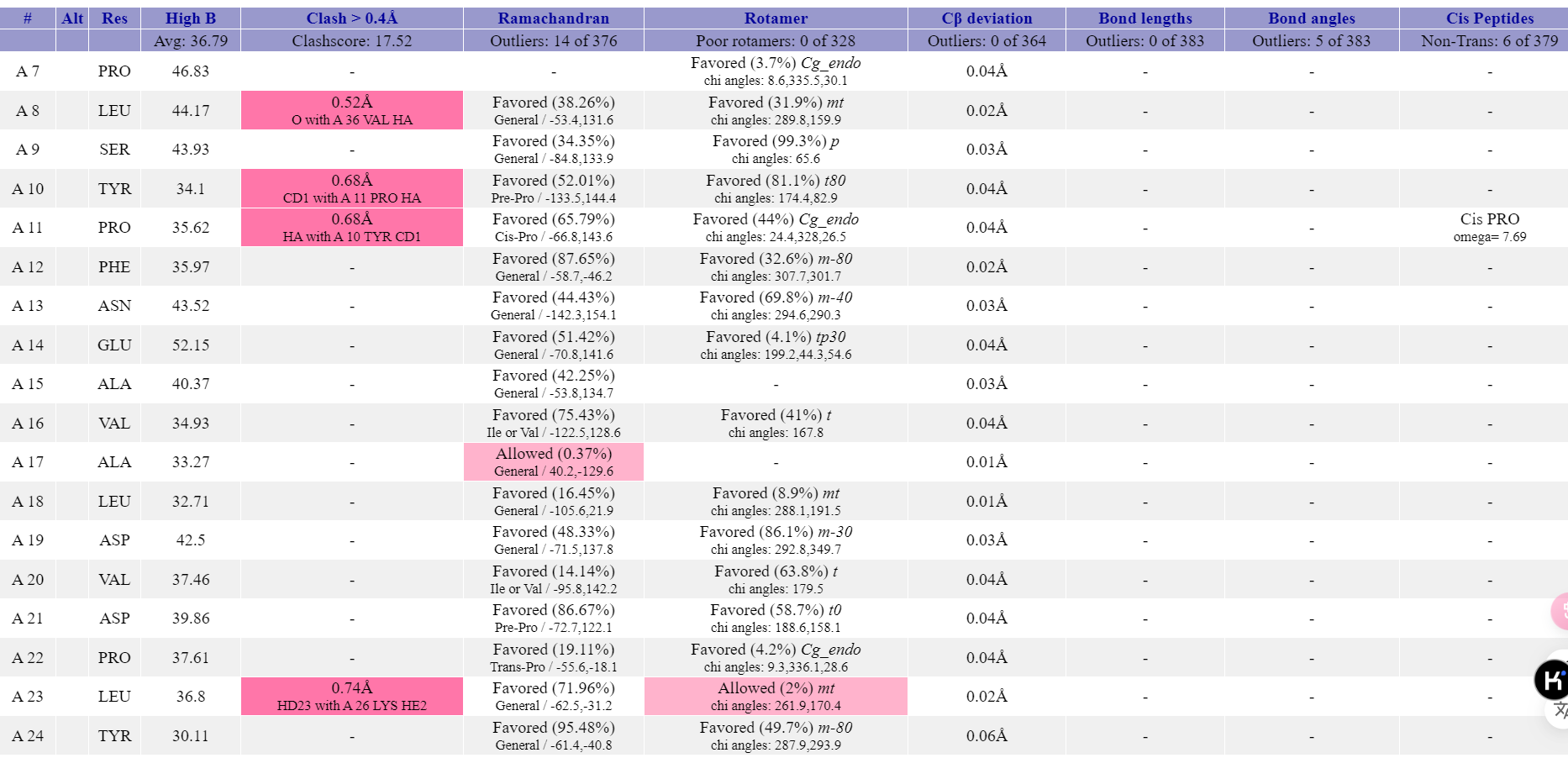
对于这些有误的地方,标红的残基需要再次打开coot进行手动精修
10,最后收集整理一下统计数据:
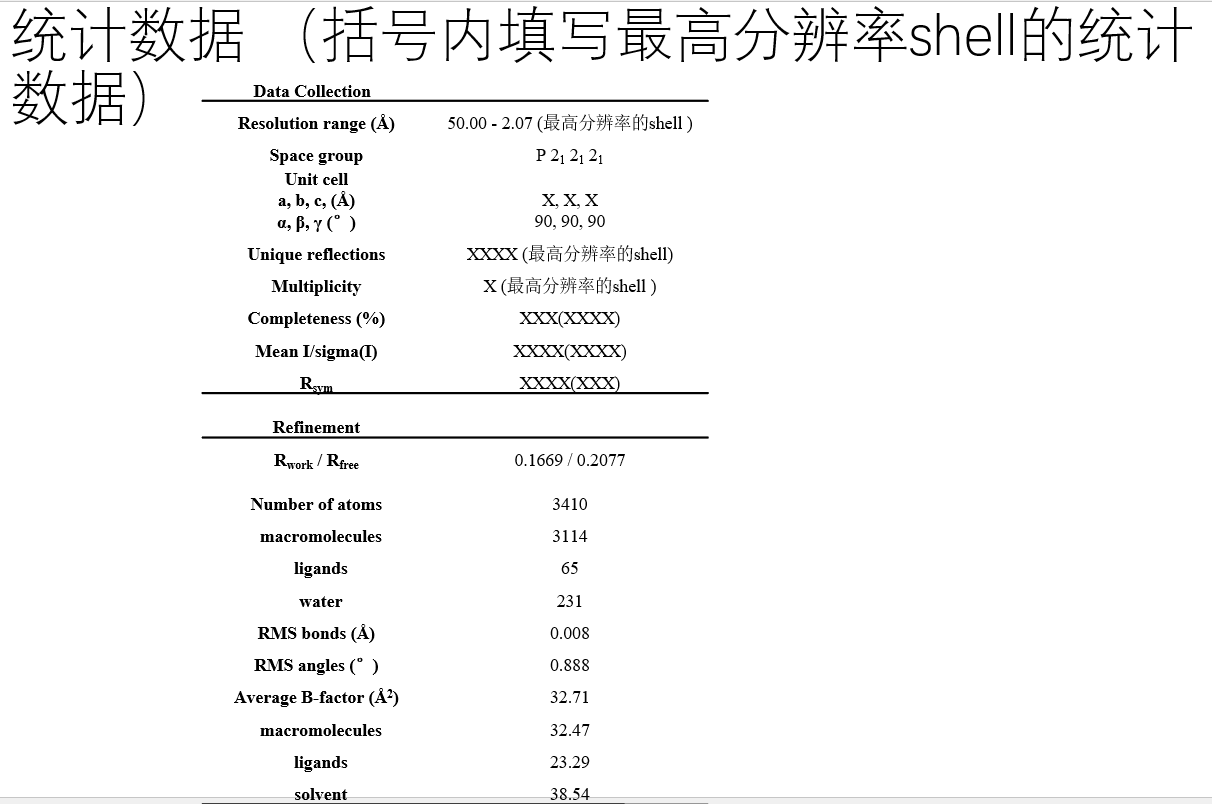
第一部分是data collection的,其实就是上一次博客的结果统计部分;
然后refinement部分是此次精修的部分,结果可以在phenix输出中找,也可以在PDB文件的表头(也就是remark中寻找)
——》然后因为使用都是同一个mtz文件,所以第一部分的结果其实就是上一篇博客的结果:
在文件correct.LP中
拉到这个文件的最后一部分:我们主要是获取分辨率最大的shell
注意是文件倒数向前最后一个的subset
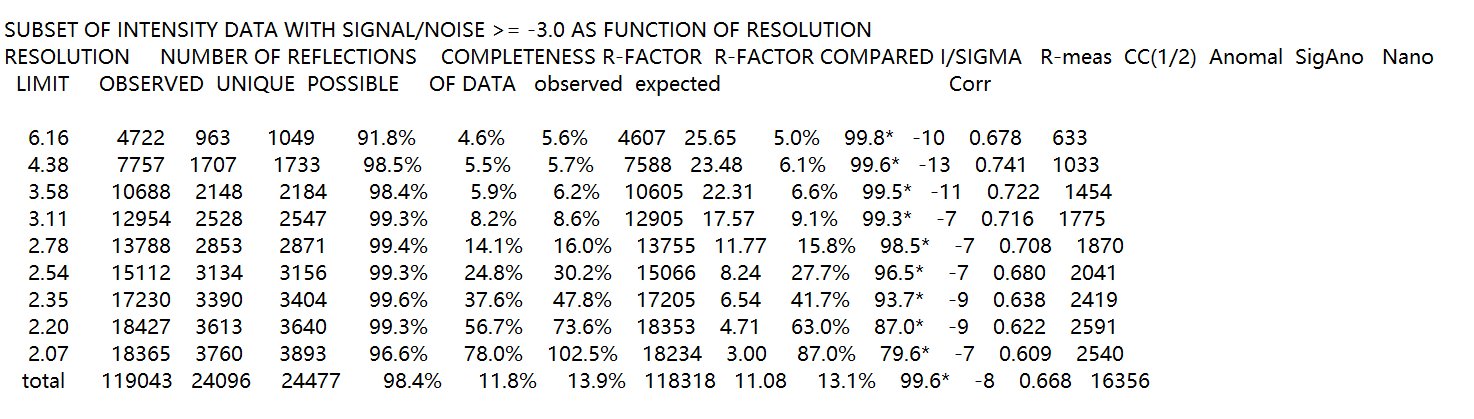 最后一列的total就是一个平均的信息,
最后一列的total就是一个平均的信息,
倒数第2行就是一个分辨率最高的一个shell的统计信息。
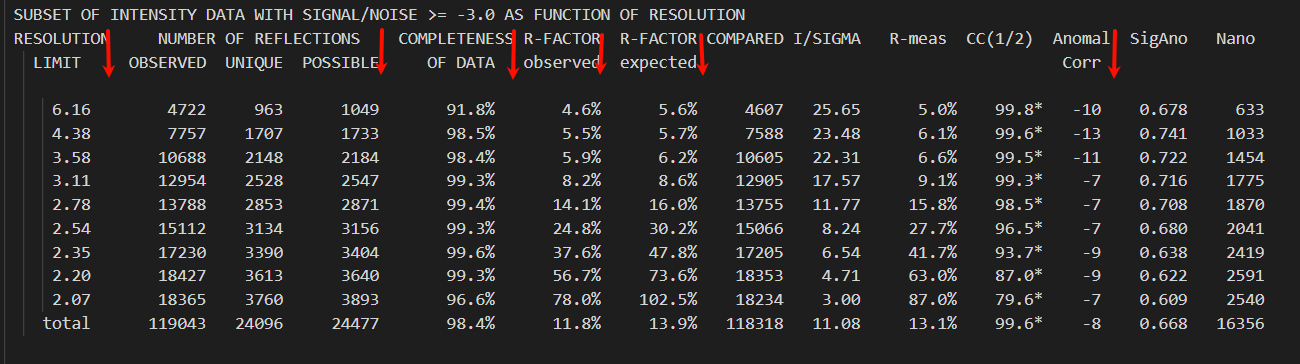
注意这里的文件的每一列的列名是占据了上下2行的,刚开始阅读时候容易把第2行也当做列名,其实是第一行中的列名写不下了;
这样竖着来看就正常多了。

















 6977
6977

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









