







承接我的上一篇博客:
https://blog.csdn.net/weixin_62528784/article/details/144698291?spm=1001.2014.3001.5501
准备好数据:示例数据见下
文件大小: 2.4 GB
分享内容: P2S14_1.7z
(数据需要上传解压7z:直接上传,或者是解压后上传整个文件夹)
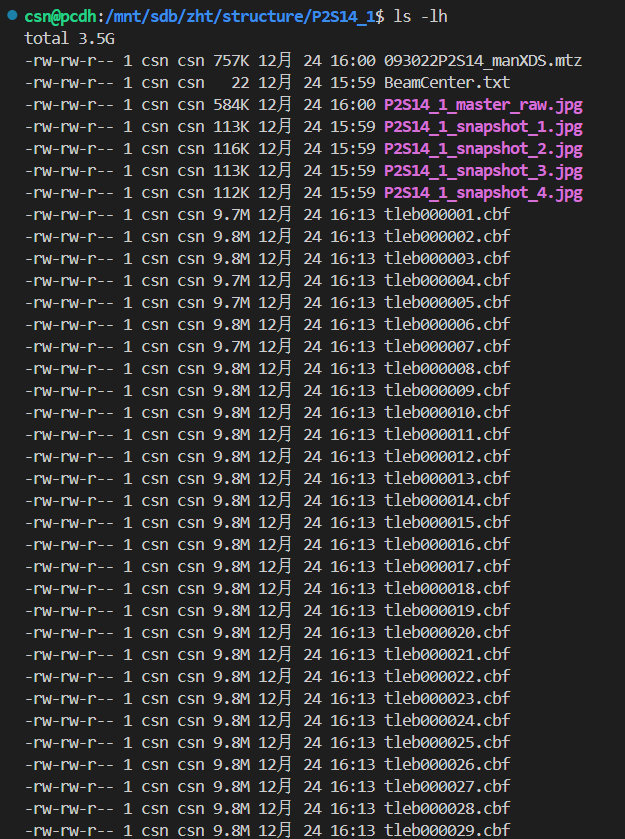
整体数据解析流程

主要进行数据索引的整合,使用工具CCP4中的iMosflm模块

目的:



问题:linux系统上安装的CCP4经常报错,windows安装的CCP4i也经常出问题,
以下操作使用windows的CCP4i2
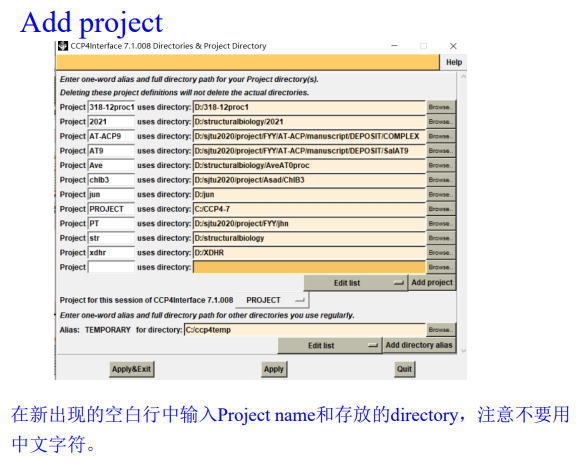
1,新建项目并打开imosflm:
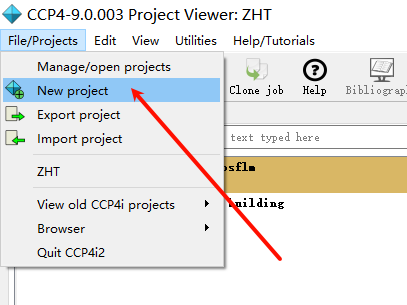
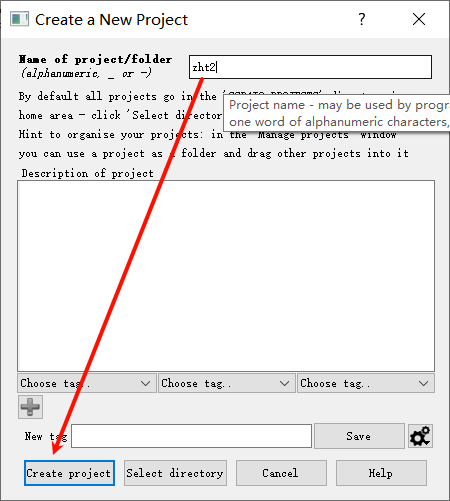
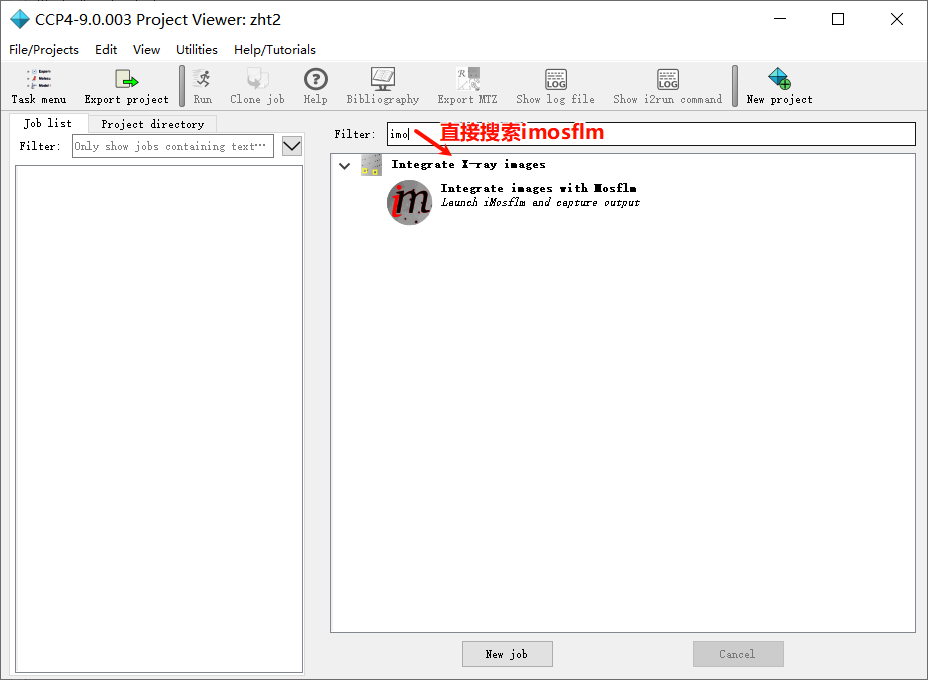
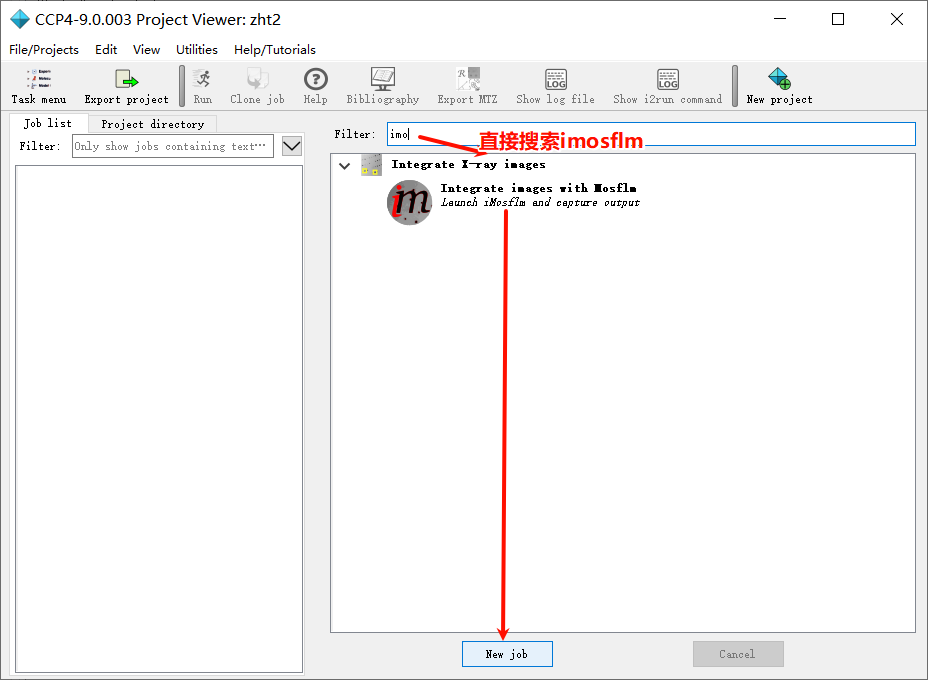
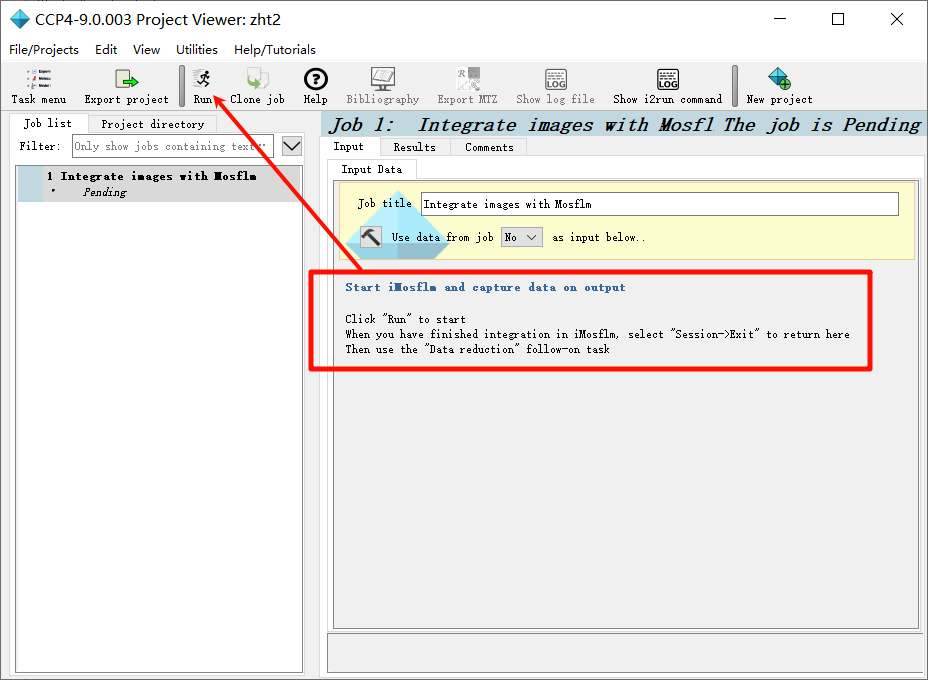
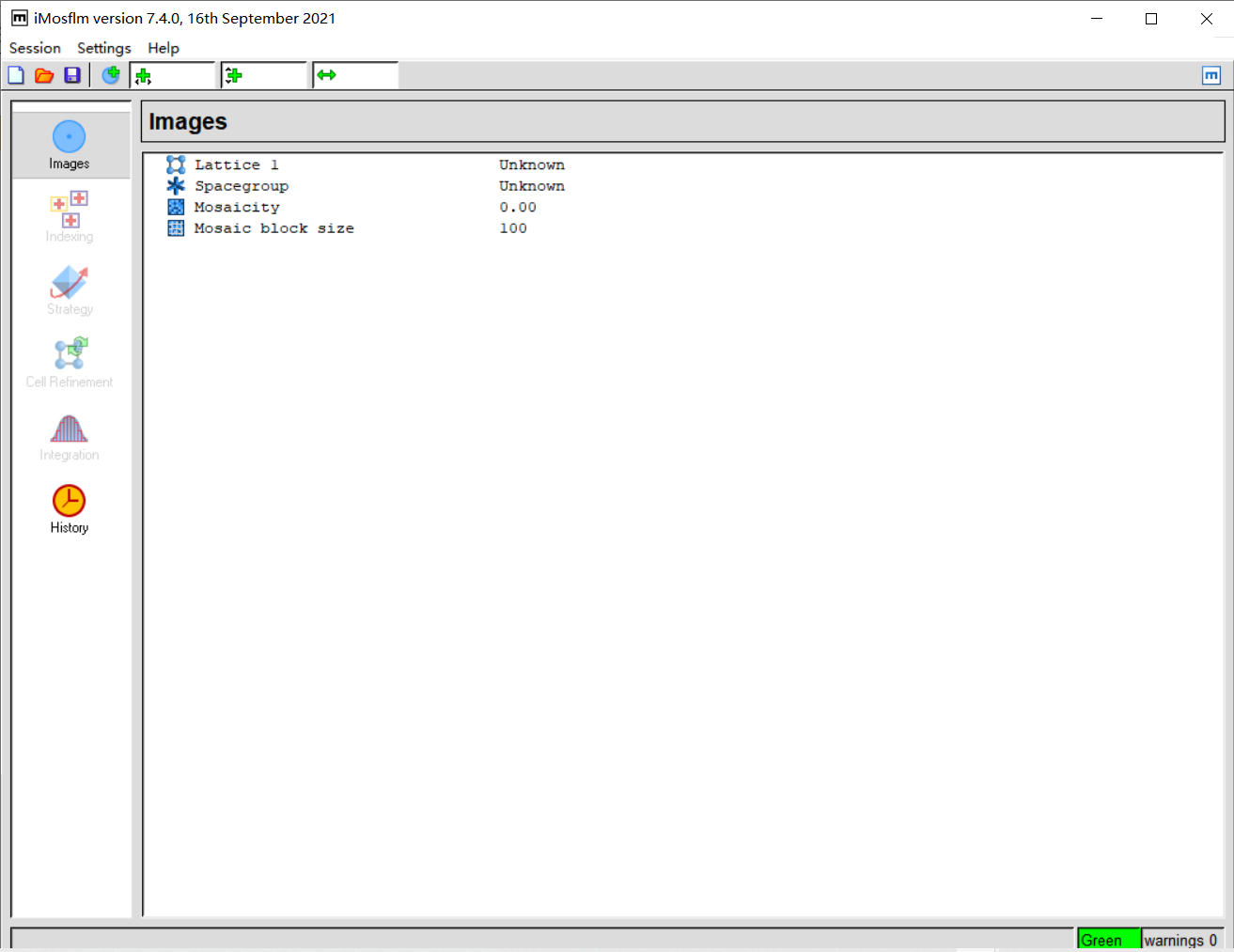
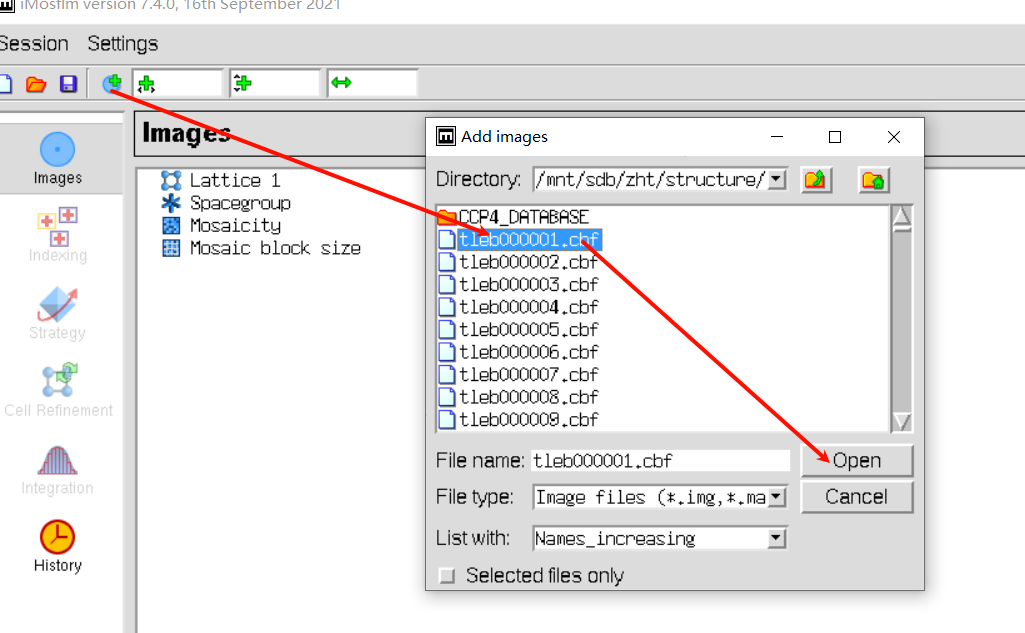
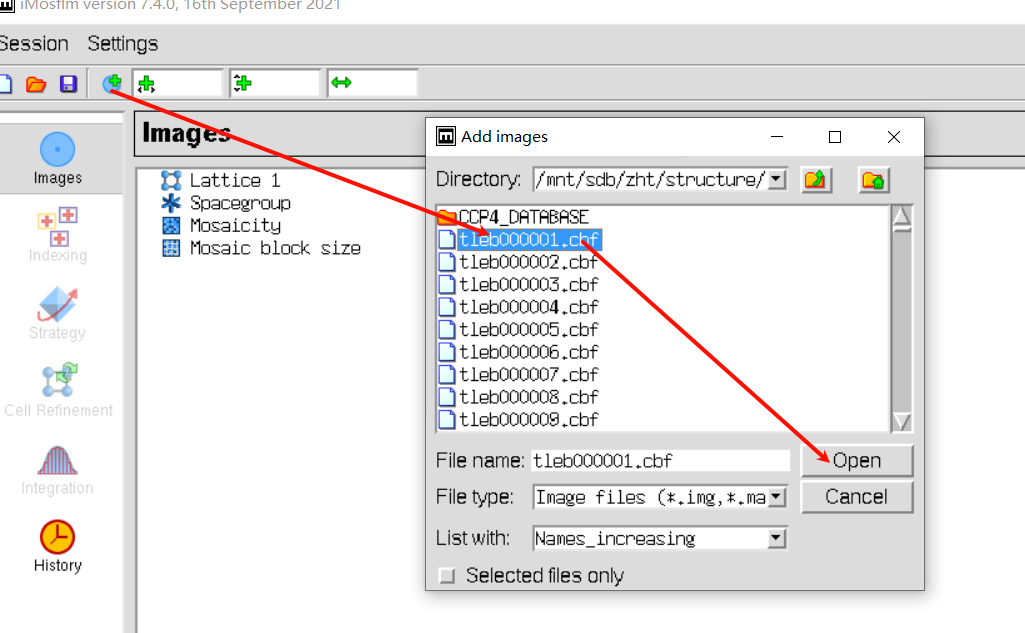
add image,在防止image的文件夹下随便添加第一张图片
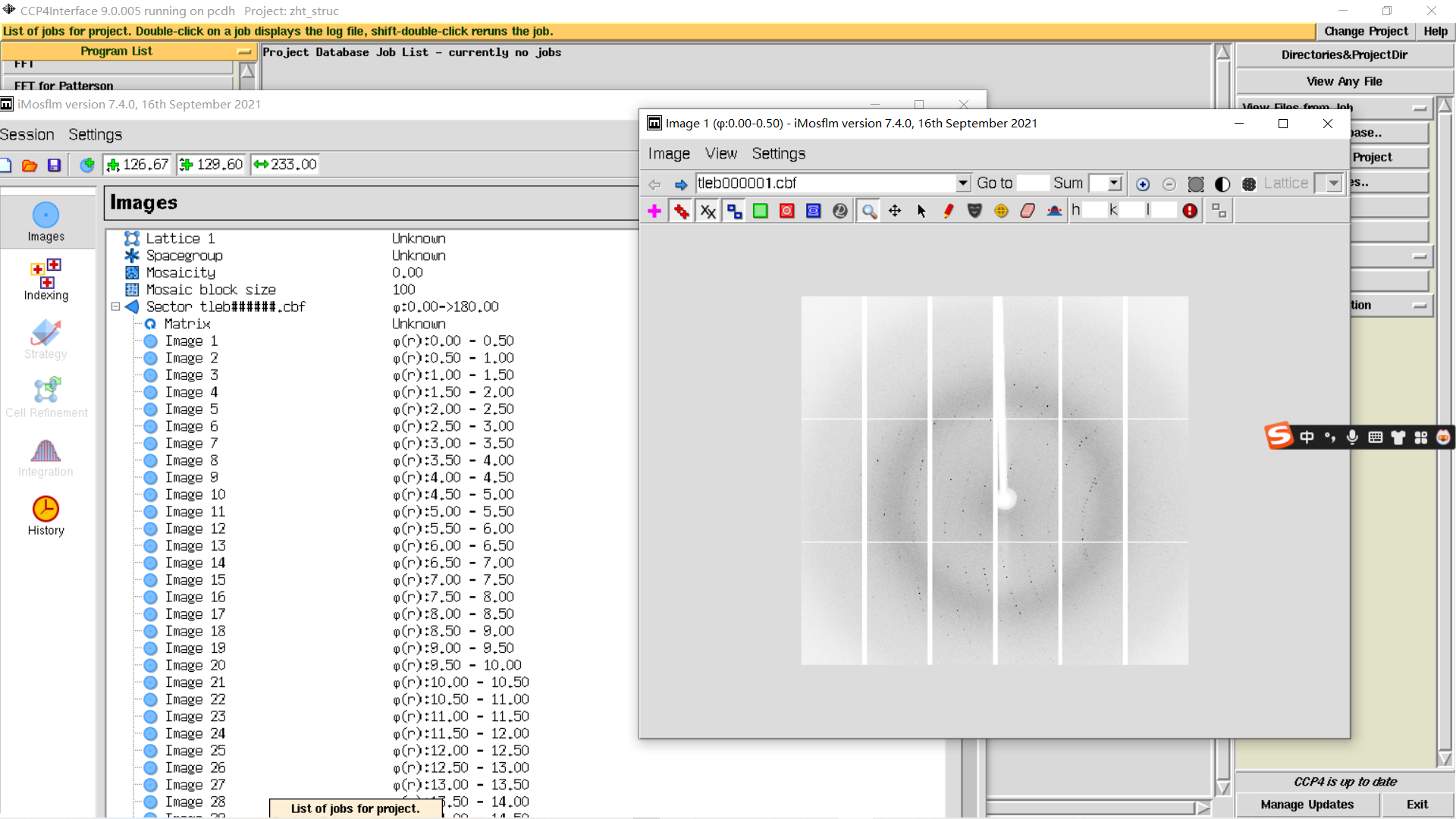
2, Contrast按钮可以调整对比度
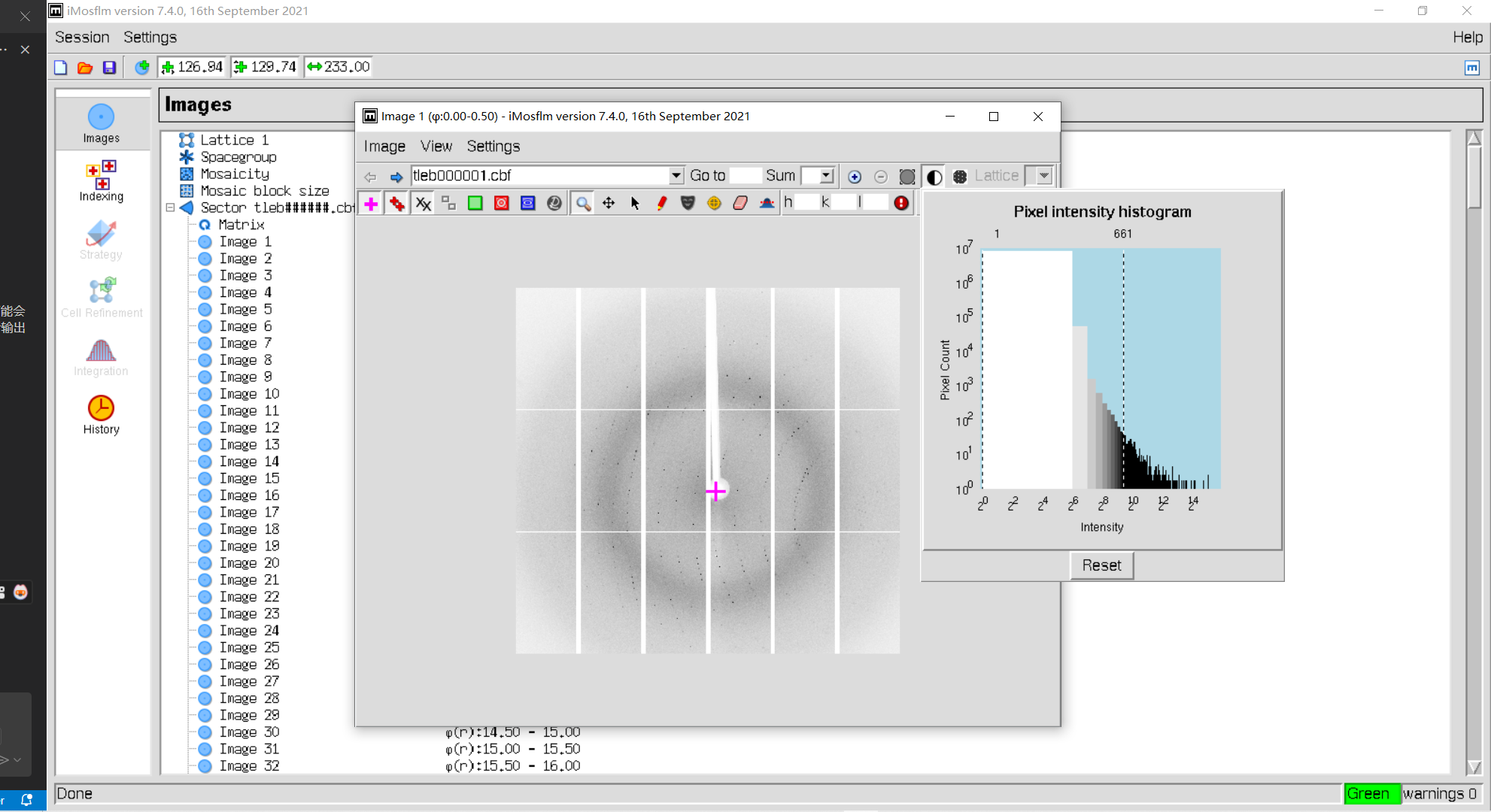
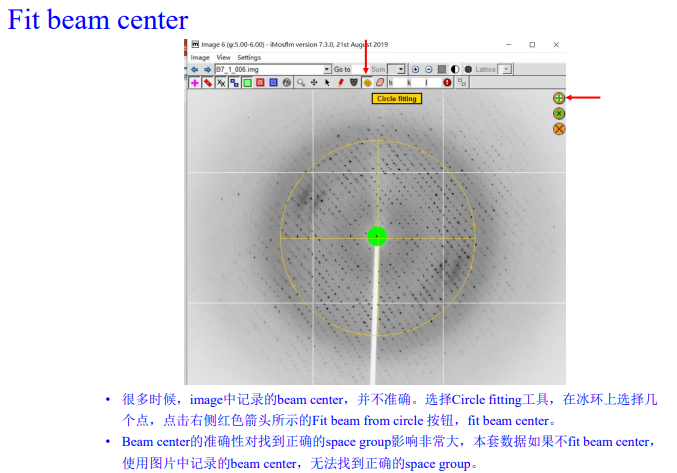
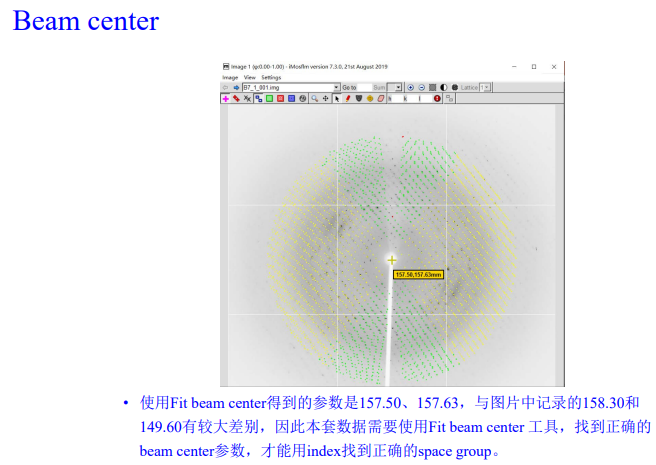
3,show beam center:
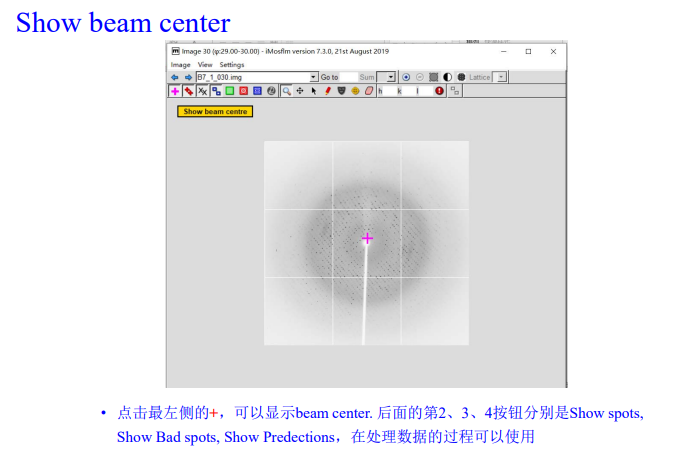
先点左上角的粉红色加号
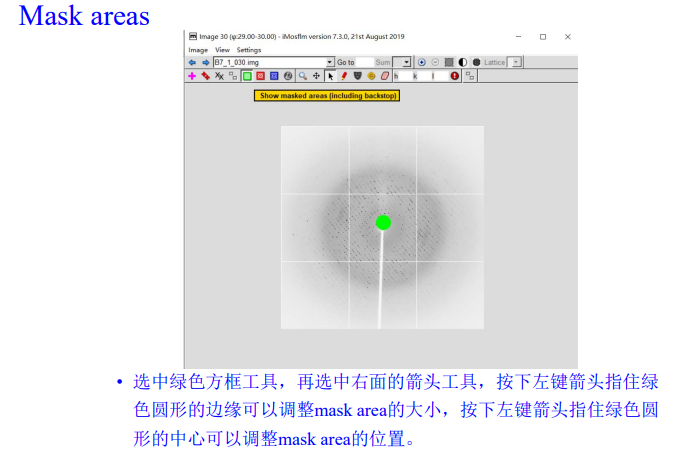
再点左上角的绿色方框,在选右边的箭头,调整大小
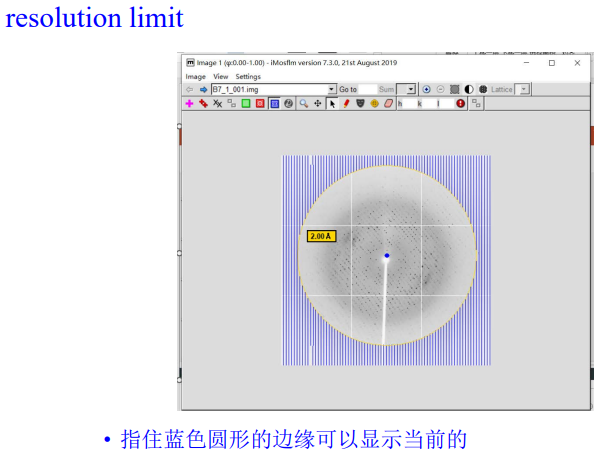
4, resolution limit & spotfinding search area
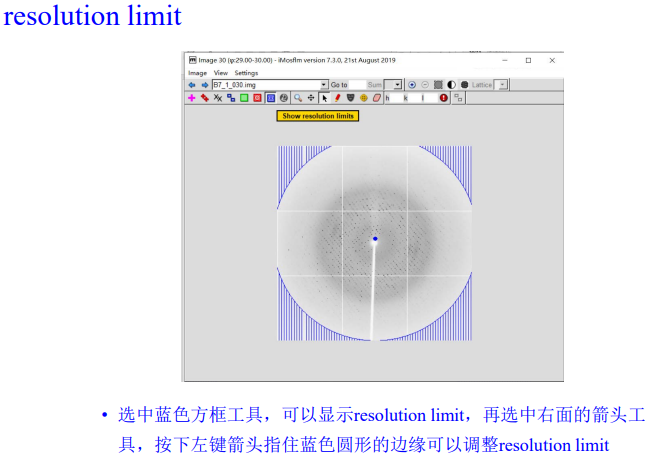
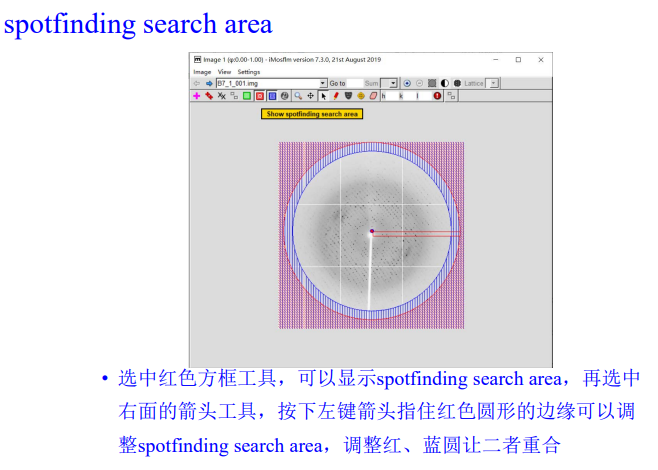
注意:
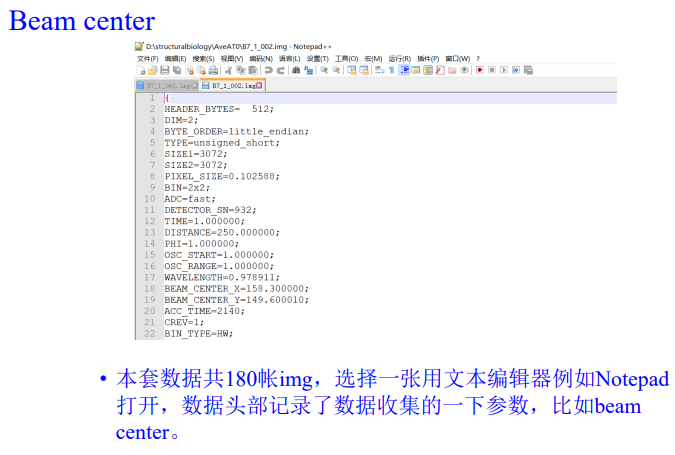
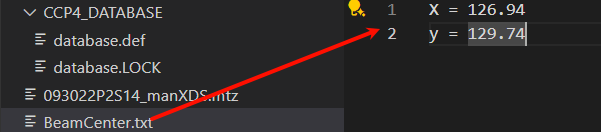
在外面这里修改:
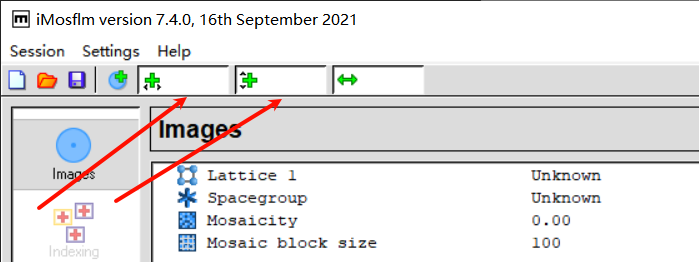

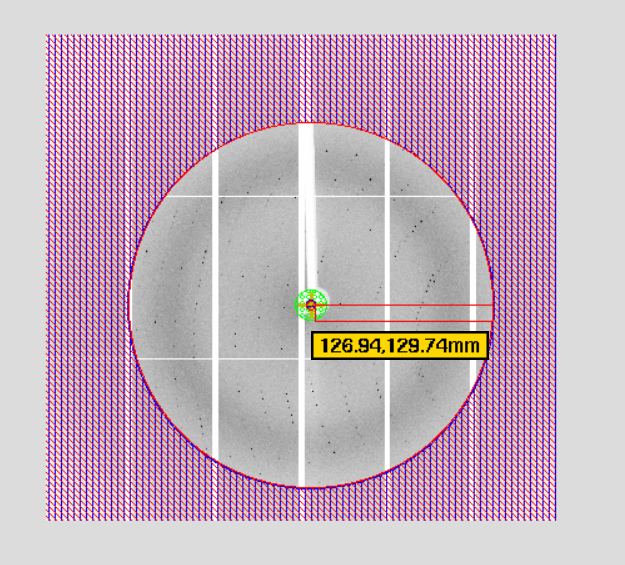
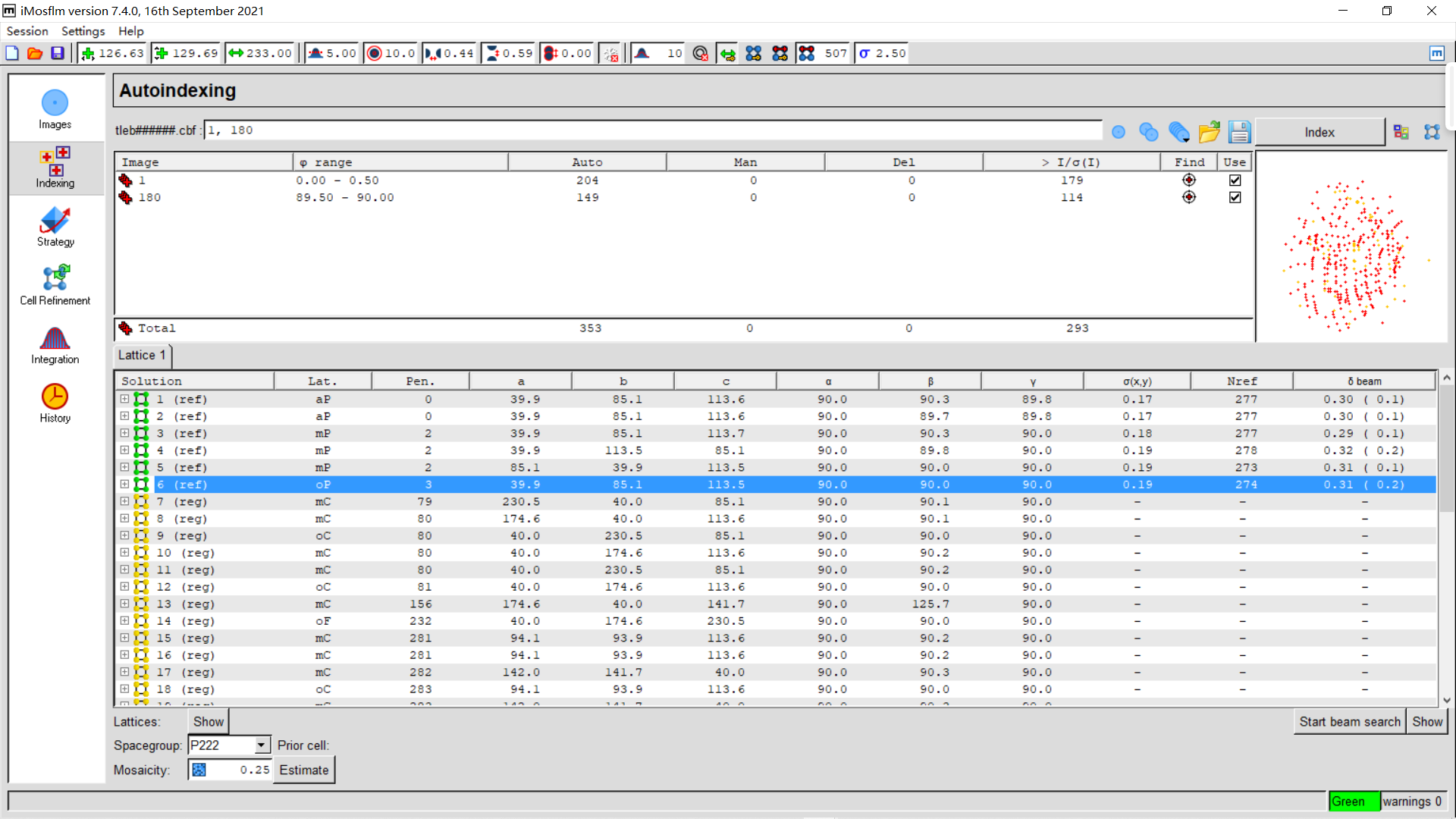
5,indexing:
从这一页可以看到晶体的空间群、晶胞参数:
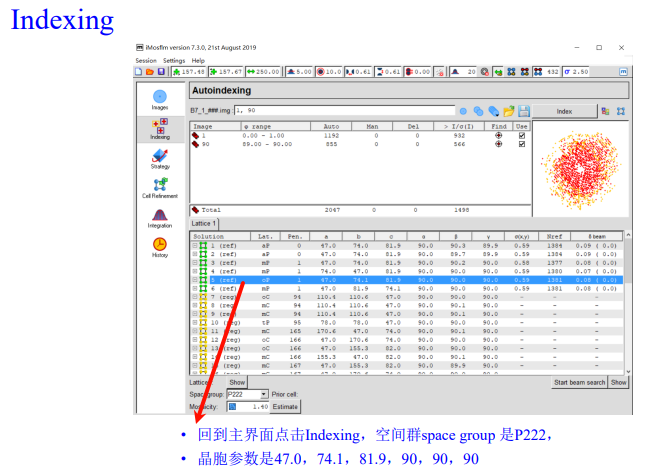

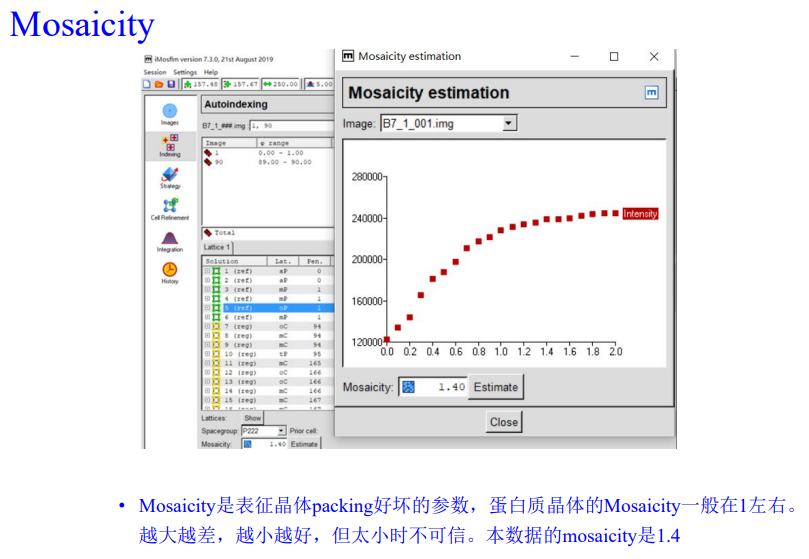
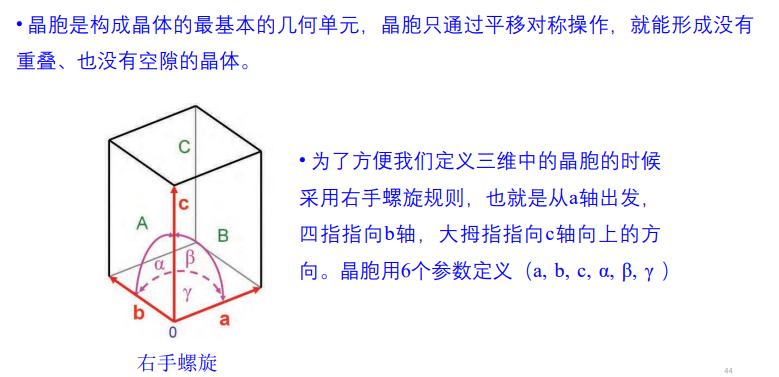
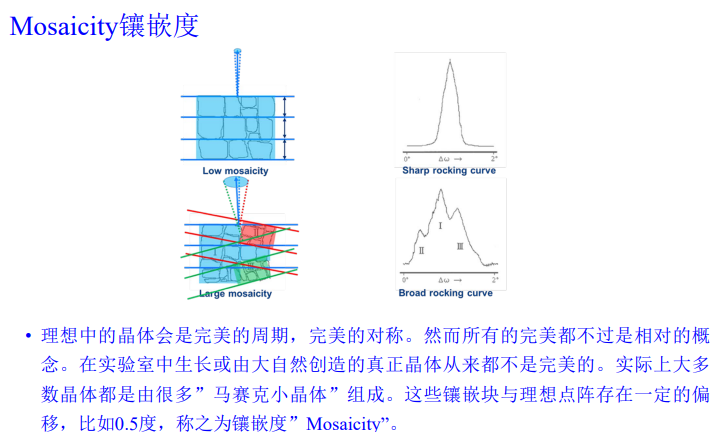
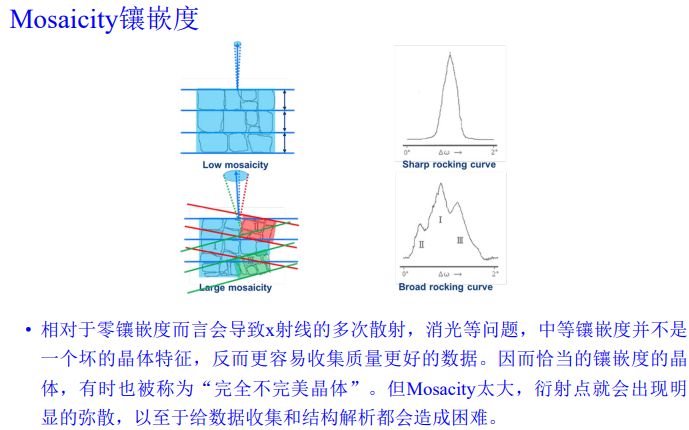
实操如下:
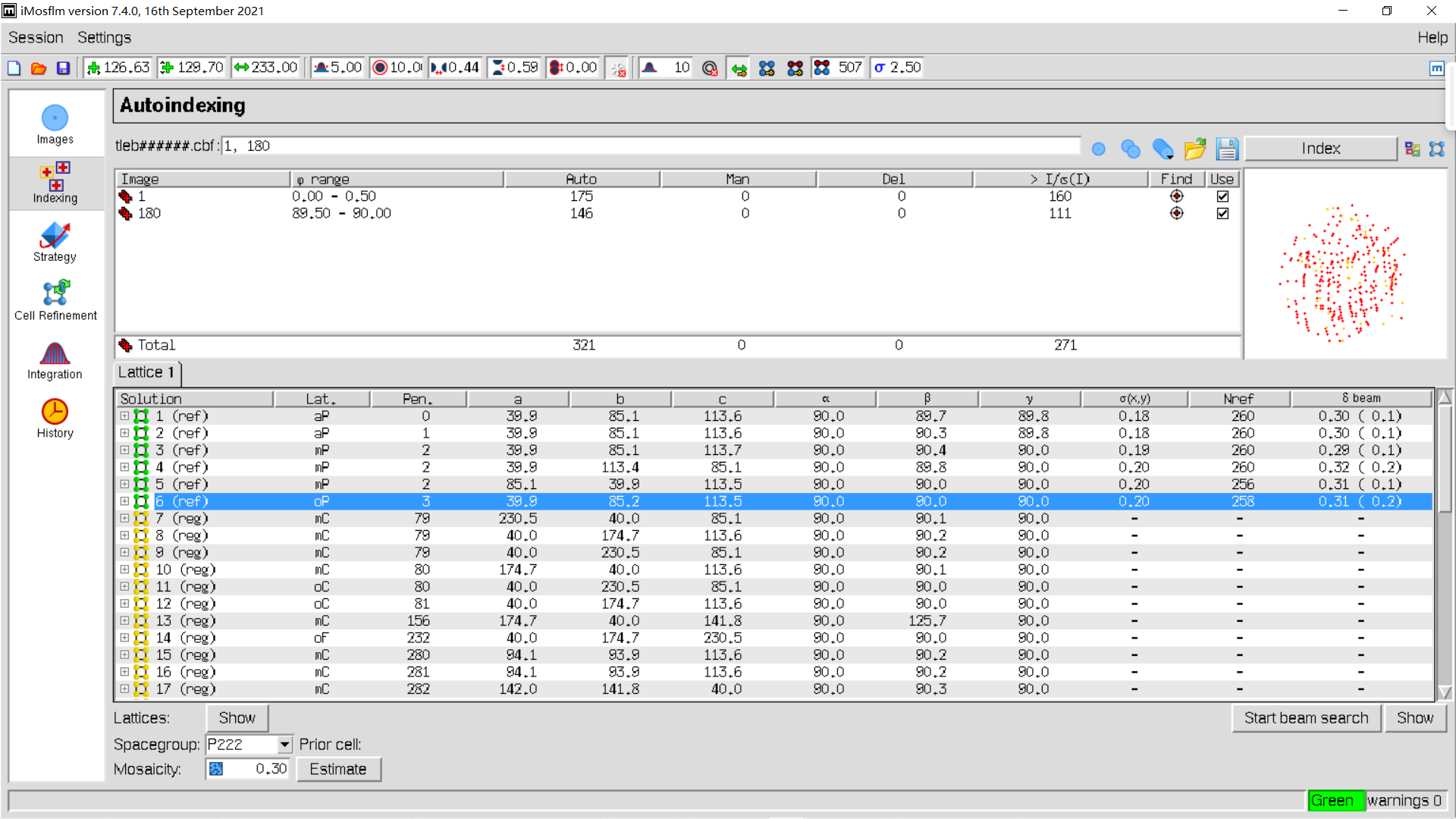
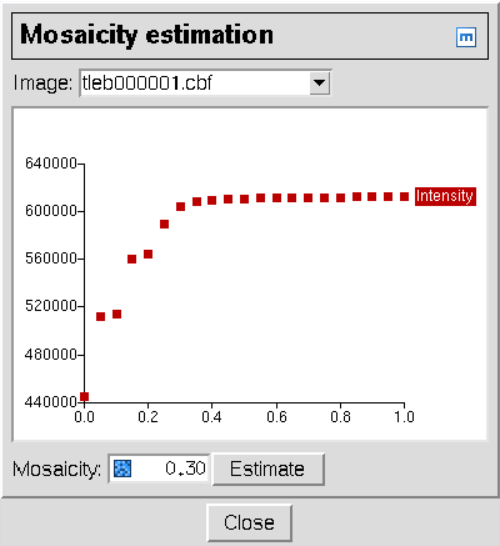
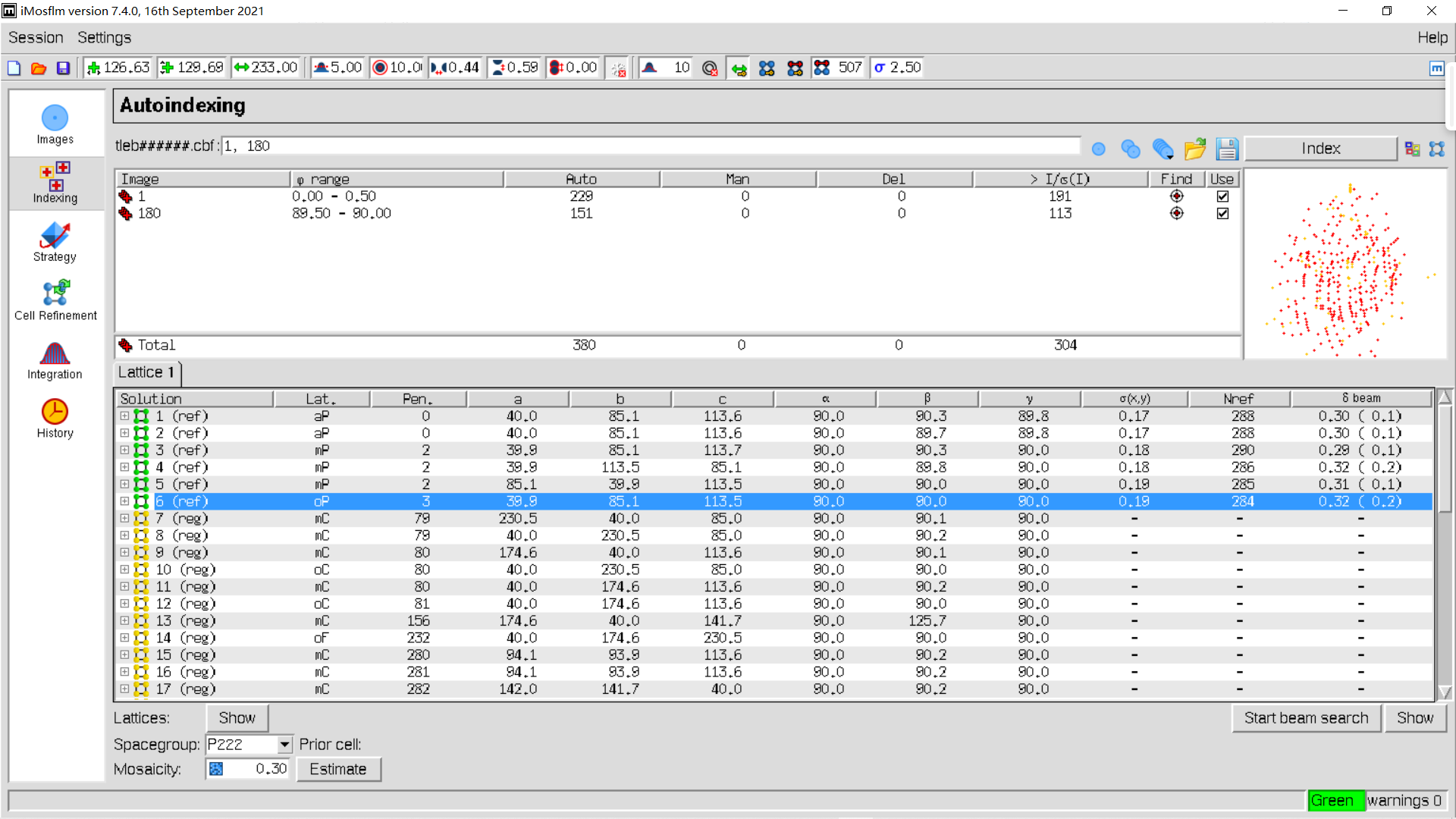
index稍作修改:
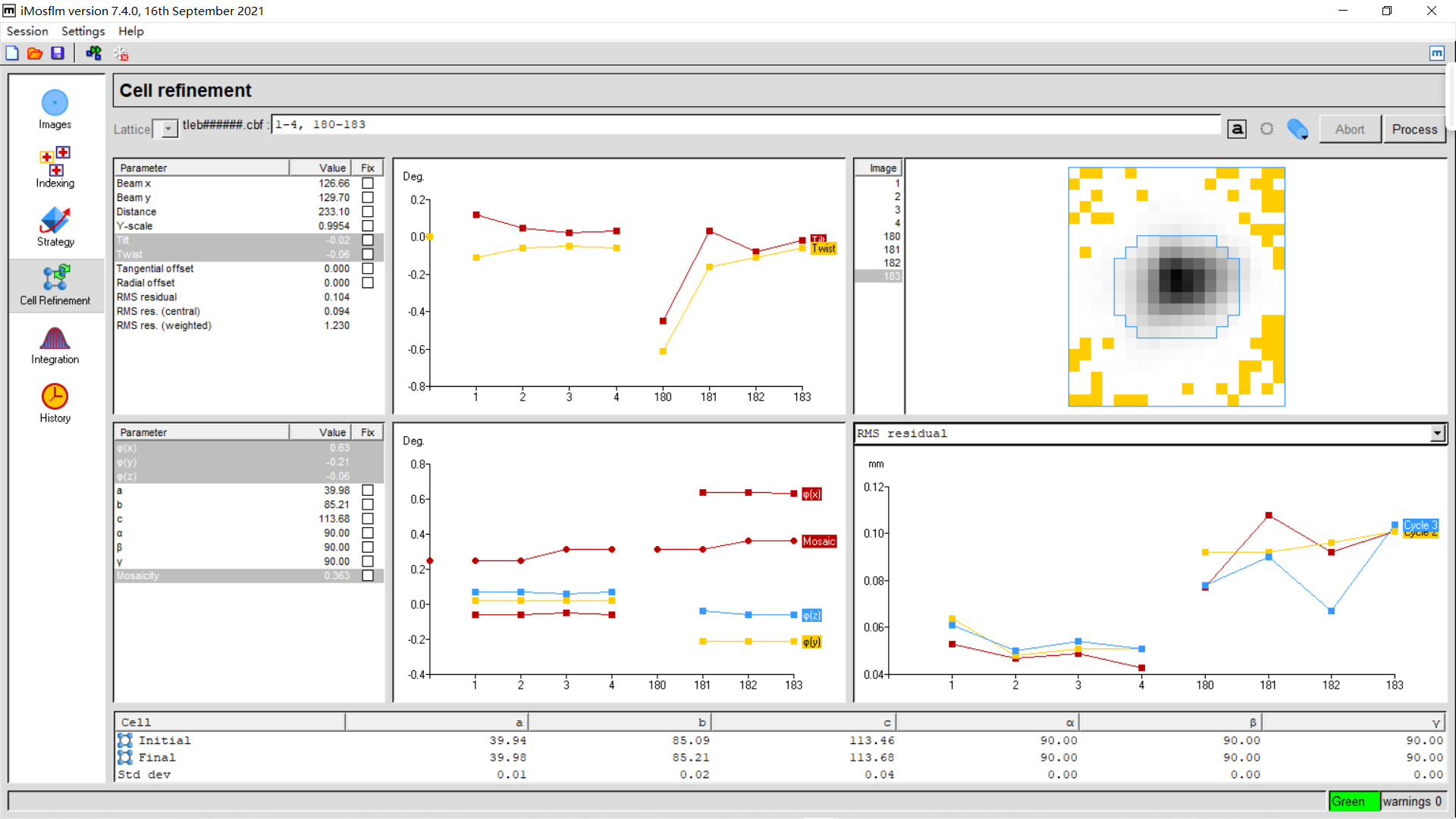
6,cell refinement:
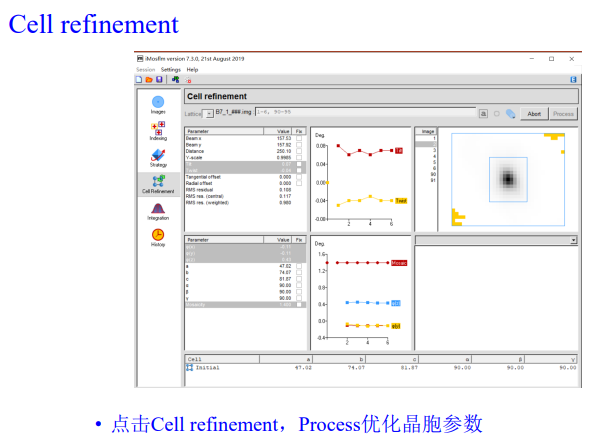
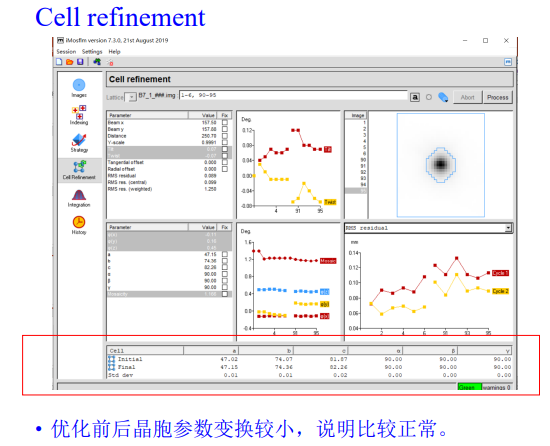
同样实操:我们可以发现优化前后晶胞参数变换较小
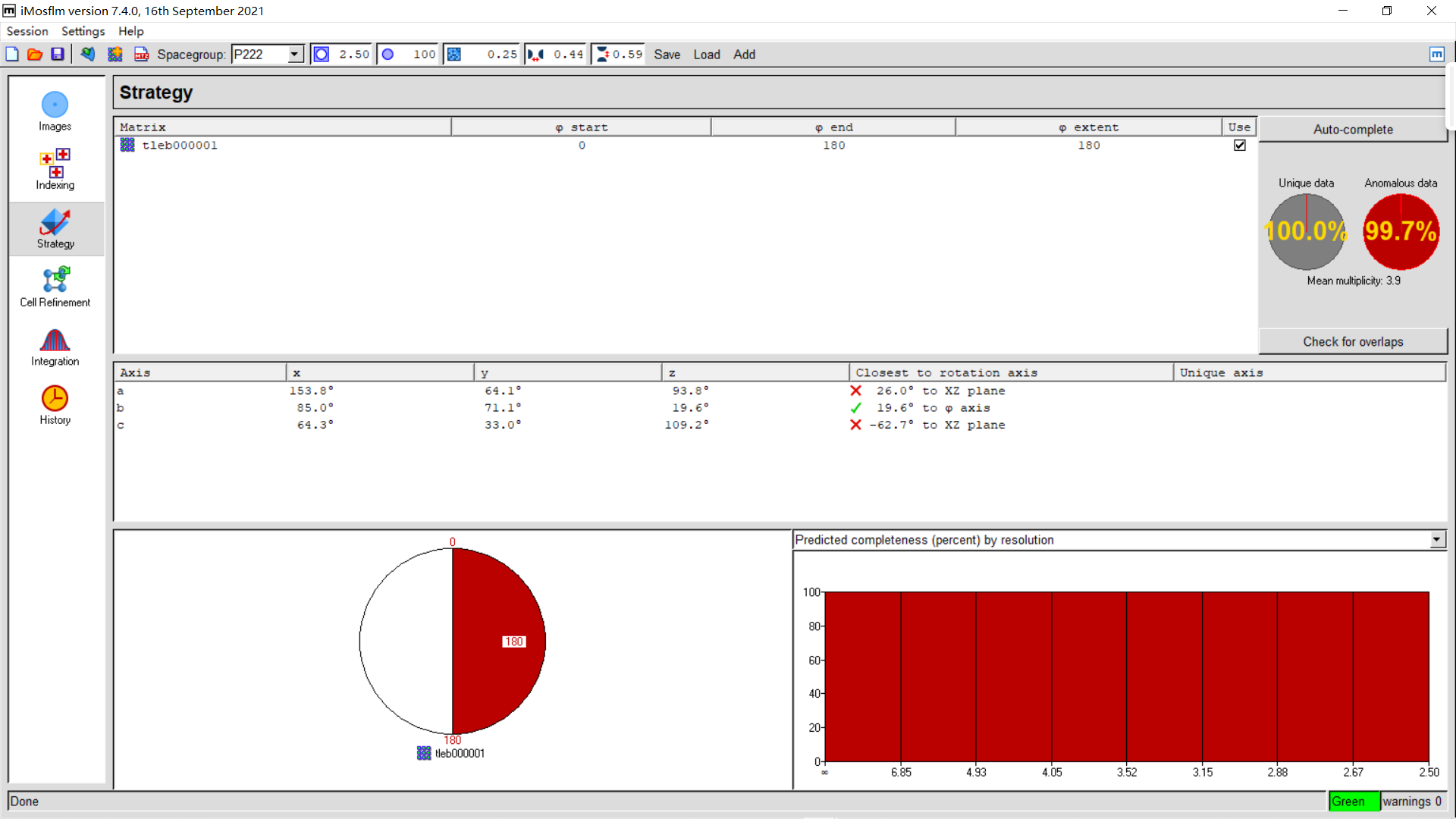
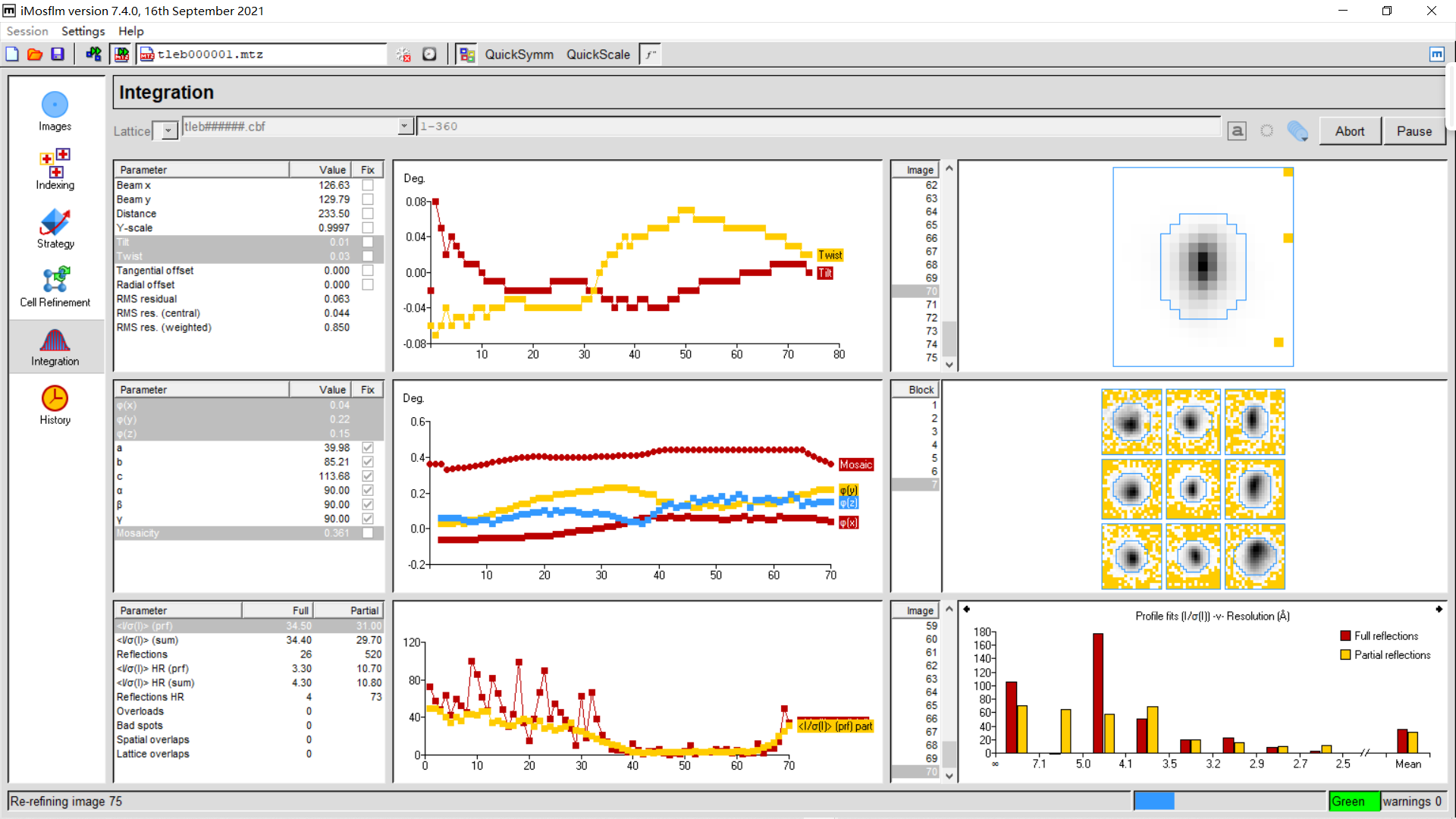
7,Integration : 点击integration、process整合数据
右上角process:
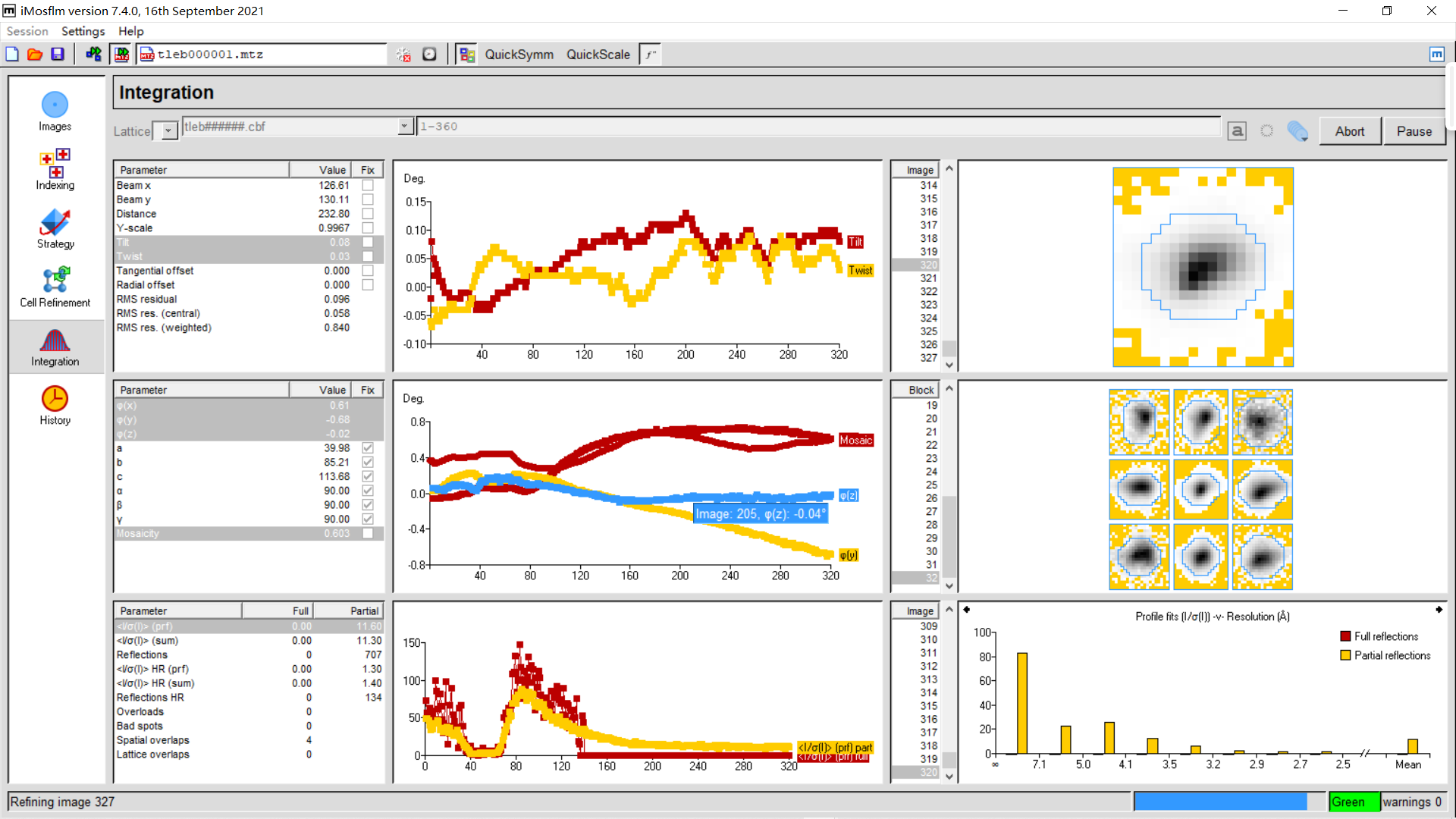
直到360张img都处理完毕:
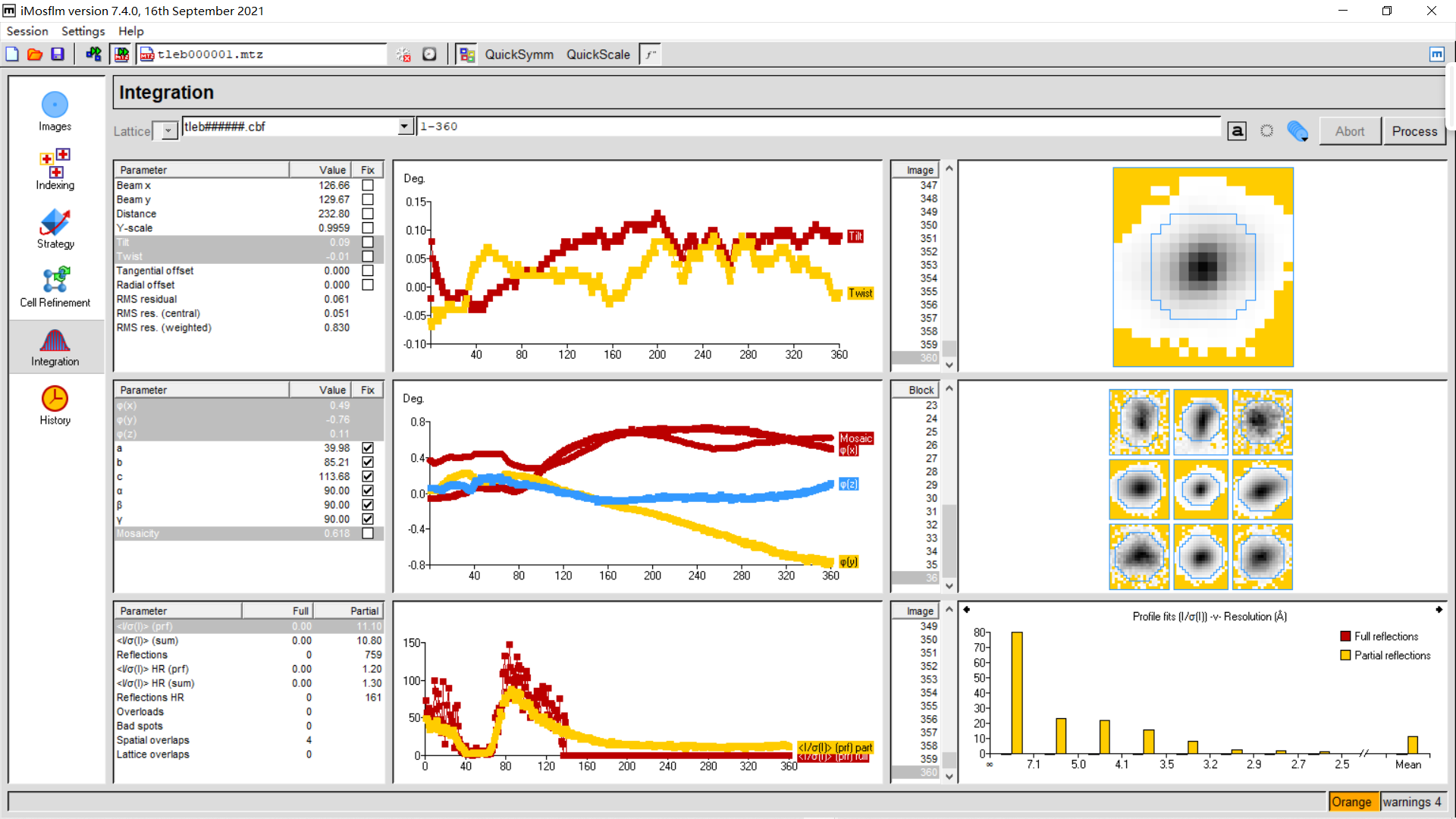
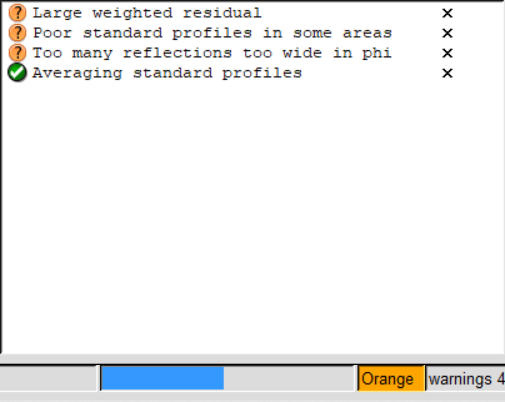
橙色还行,有点小问题但还能处理
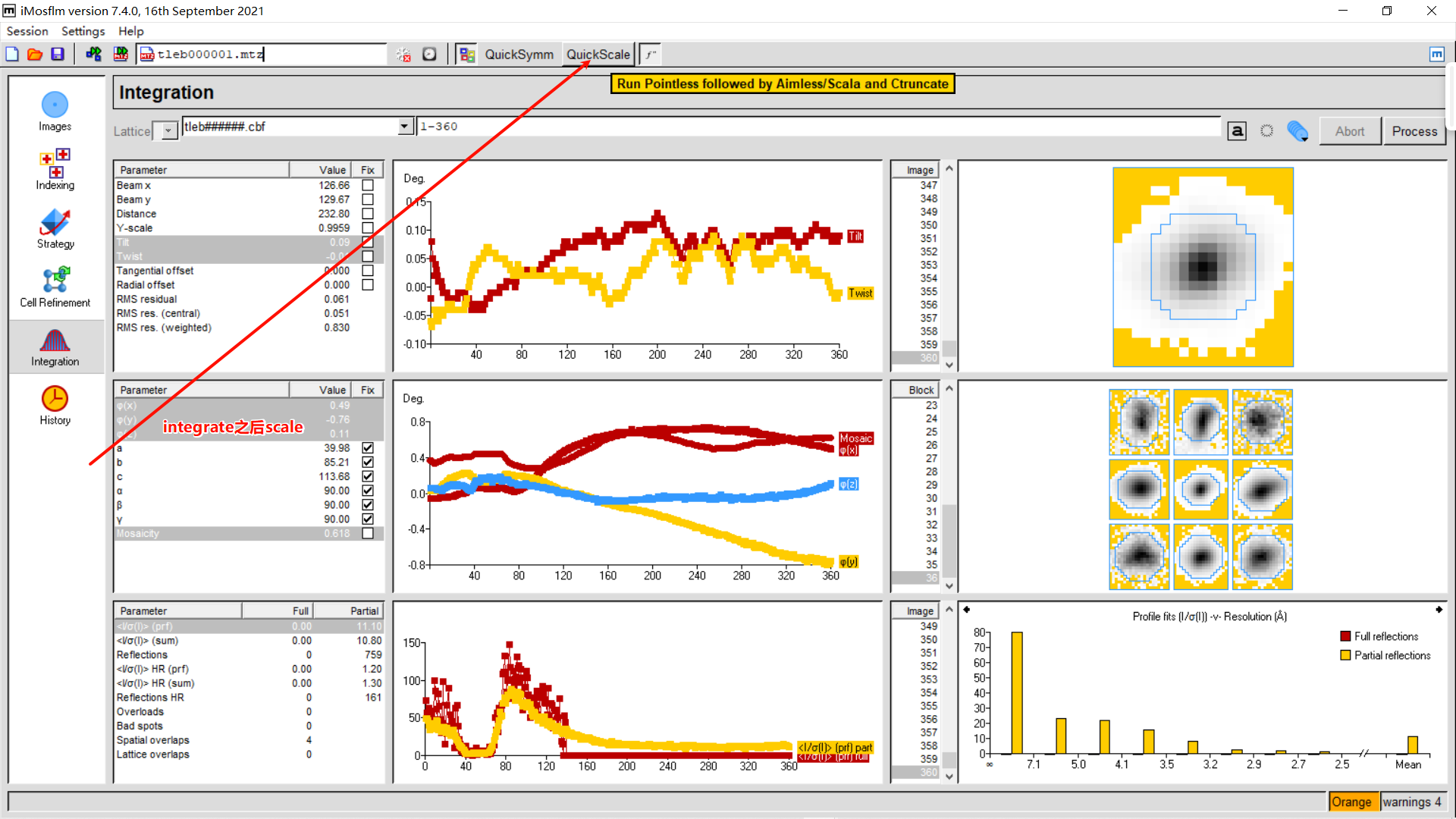
8,quick scale:
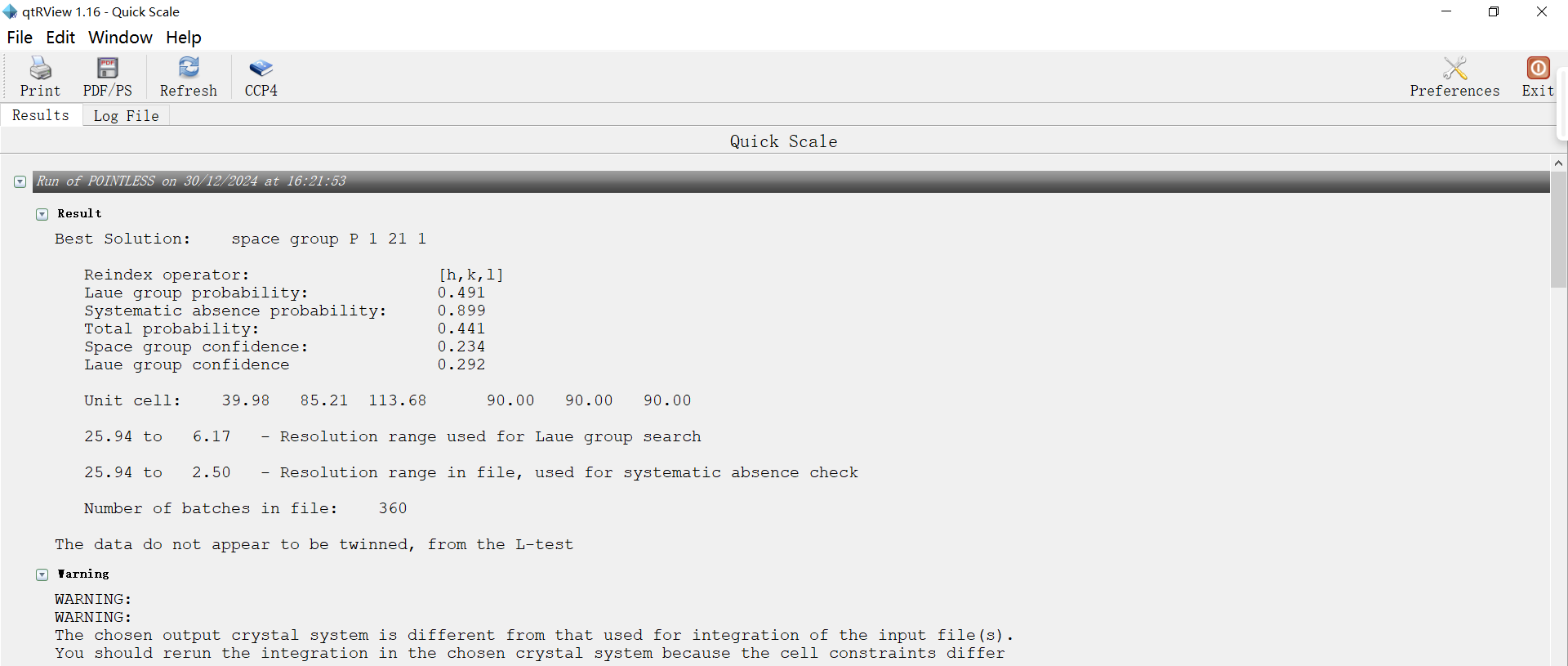
tleb000001.mtz是integrate之后的文件,每个点是分开计算的,我们应该会在viewHKL中看到大约十几万到几十万个reflection,而merge&scale之后,相同的点会被记录为一个,进行平均、求标准差的过程,这个mtz是我们需要的mtz,而这个时候的reflection 大约应该是几万个,被称为Unique Reflecion(冗余度=总衍射点数/Unique Reflection number)。如果用tleb这套数据(最前面的原始数据),可以在imosflm integrate之后点击quick scale,生成scale后的mtz文件,(ctruncate_tlebXXX.mtz),然后观察

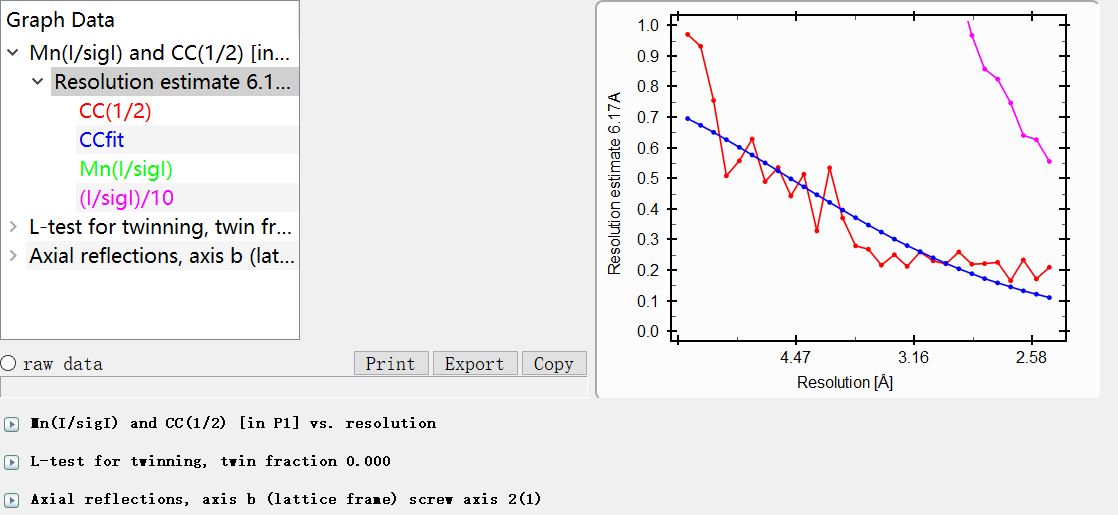
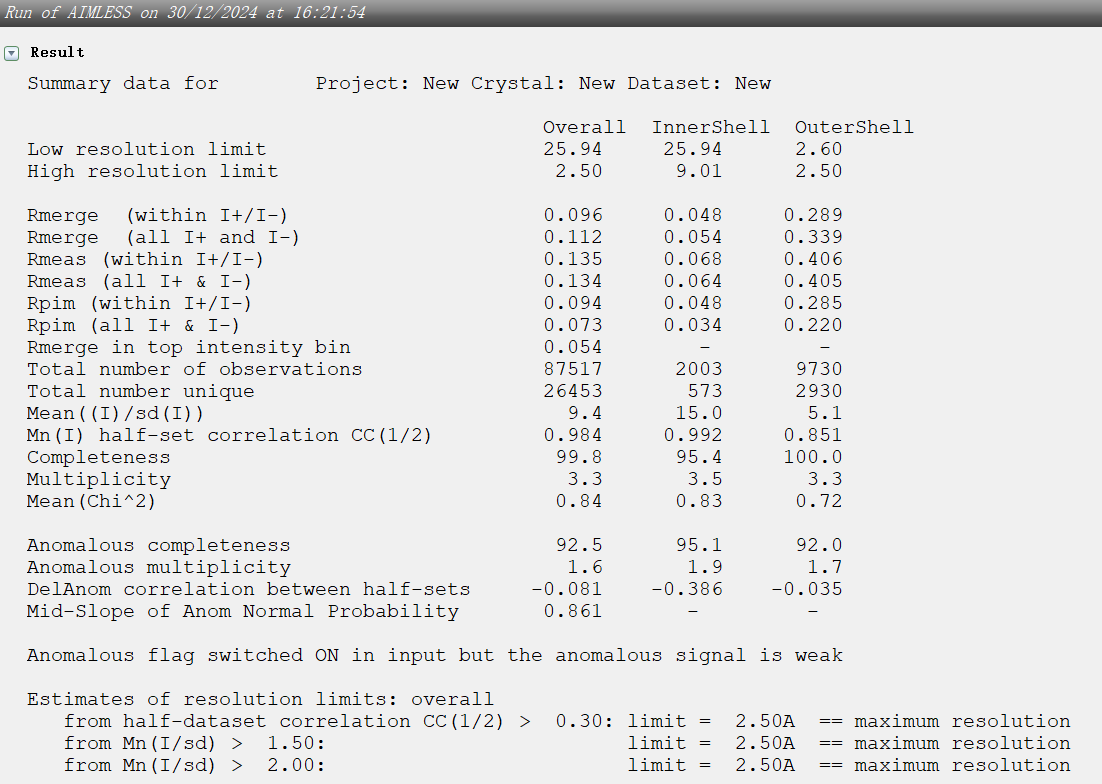
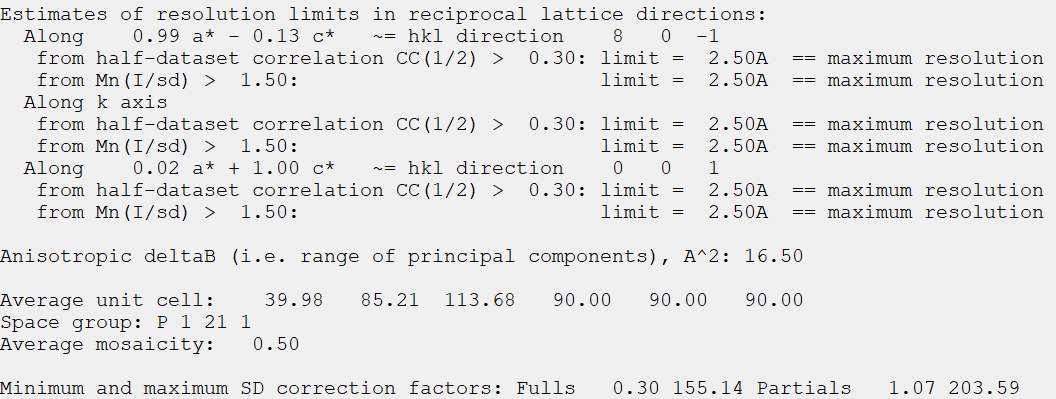
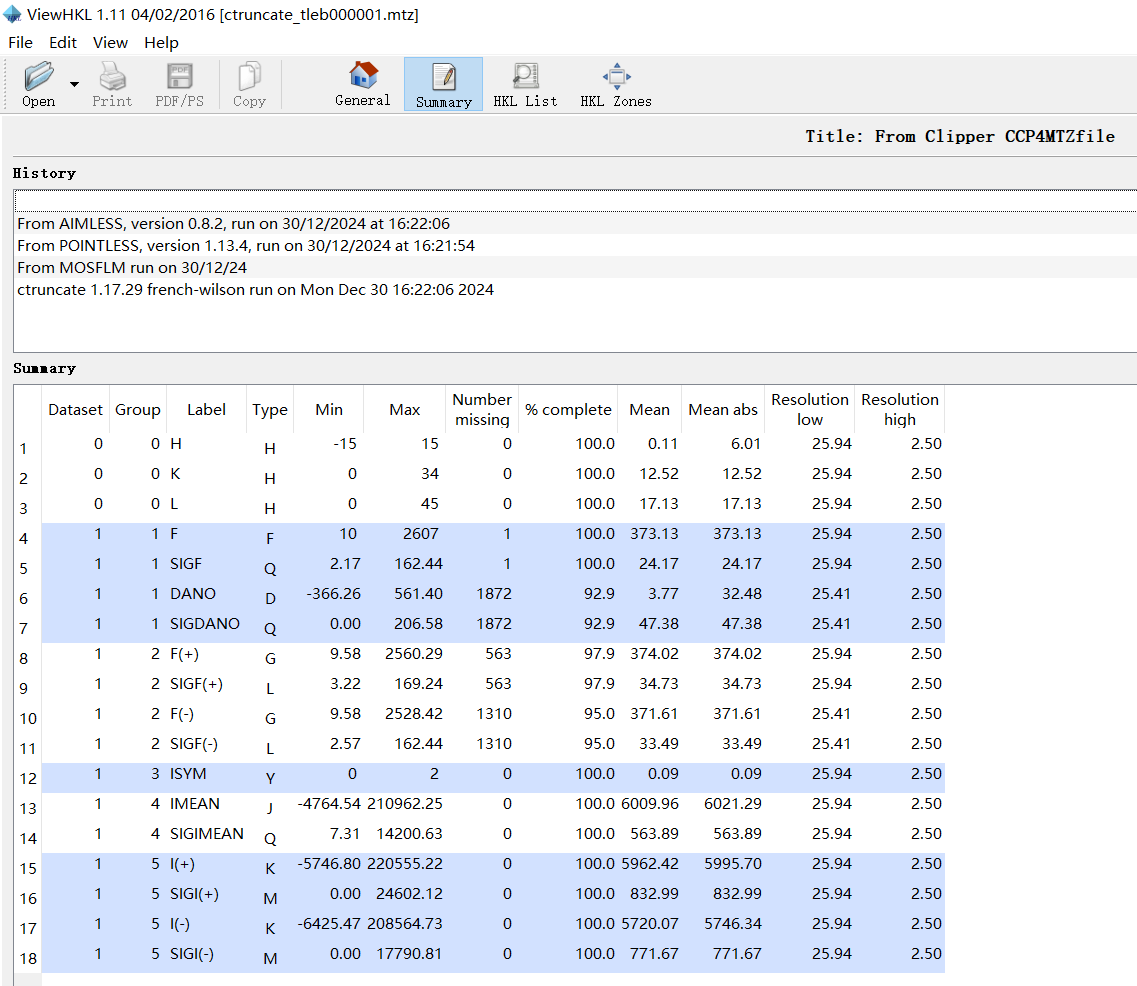
9,viewHKL是scale&merge之后的mtz文件数据:viewhkl观察数据
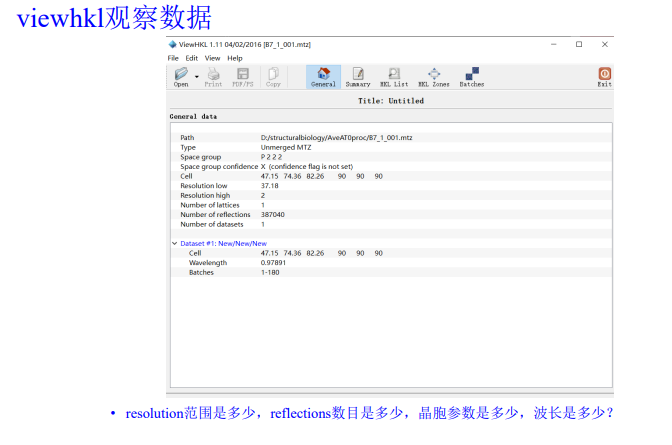
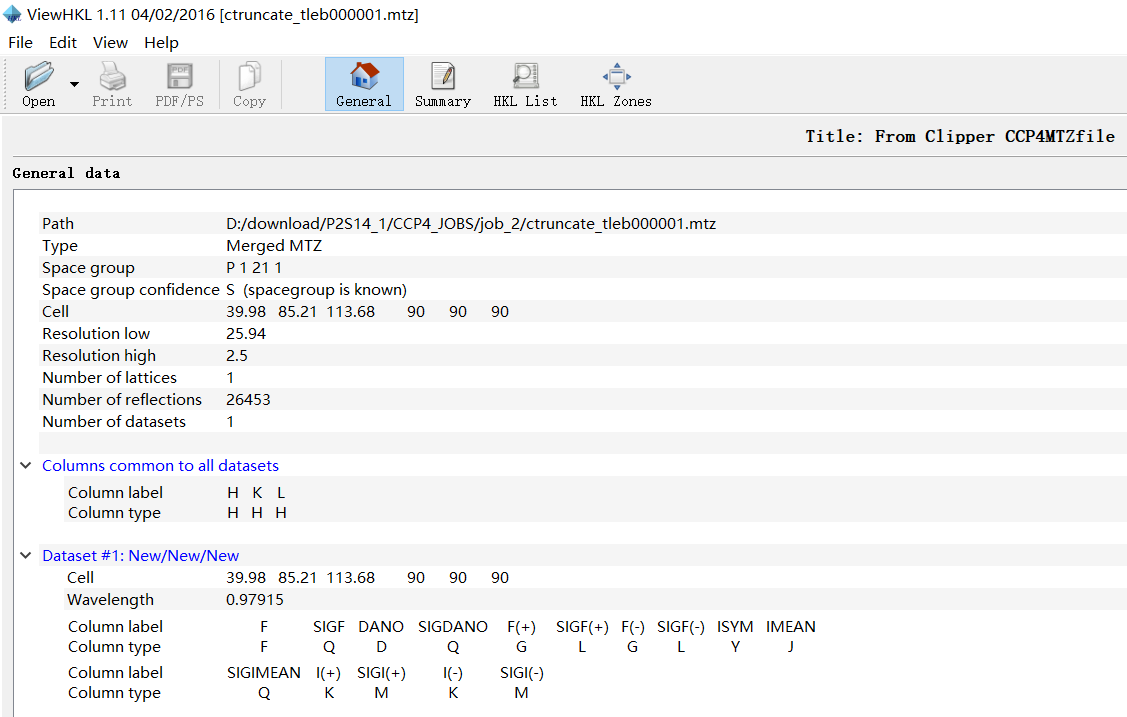
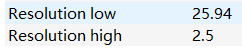
resolution范围:
reflections数目:
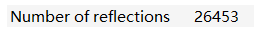
晶胞参数:

!!!!!!!!!!!!!!!!!!!!!!
过程草稿:主要是linux系统上的操作记录
!!!!!!!!!!!!!!!!!!!!!!
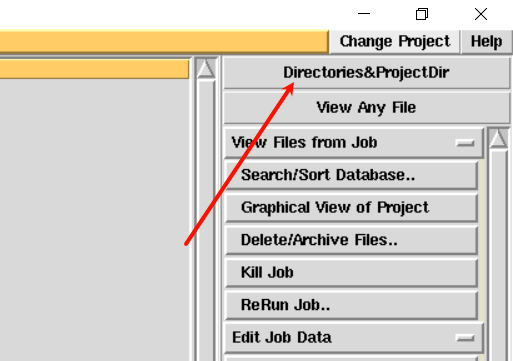
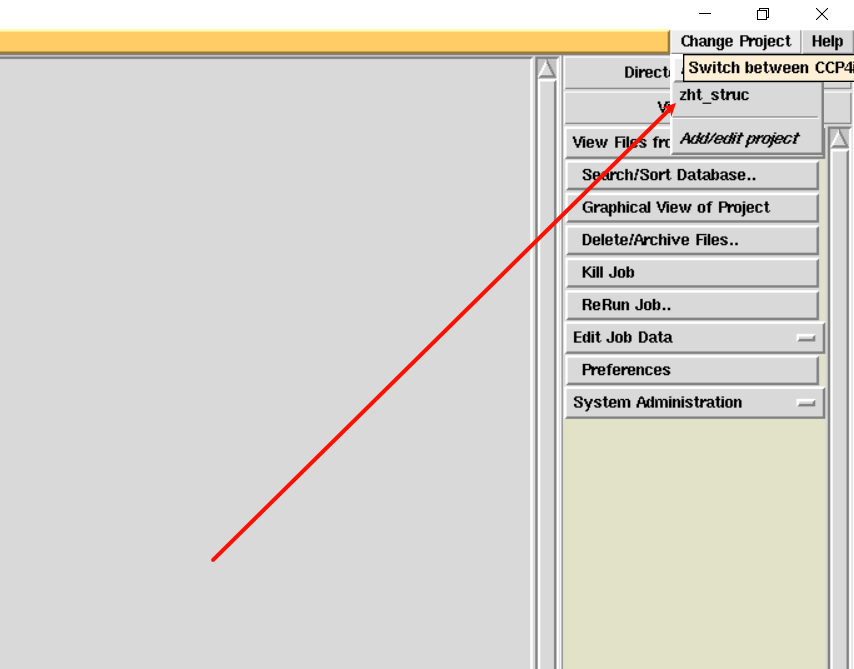
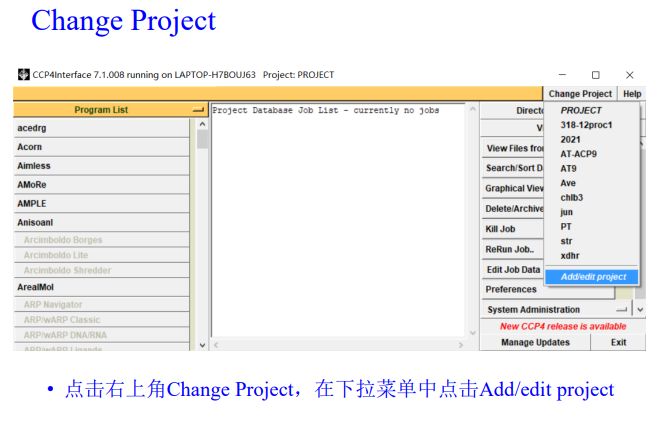
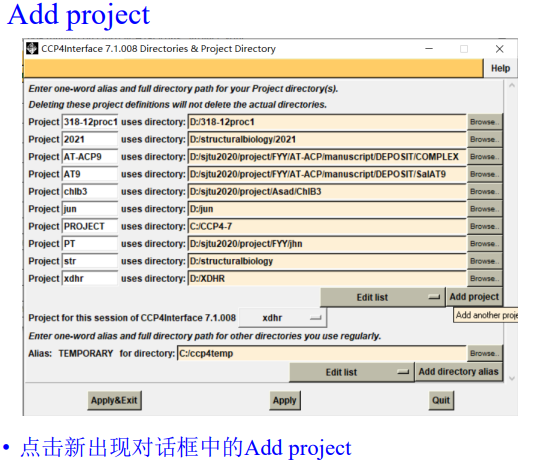
2,首先是新建项目:
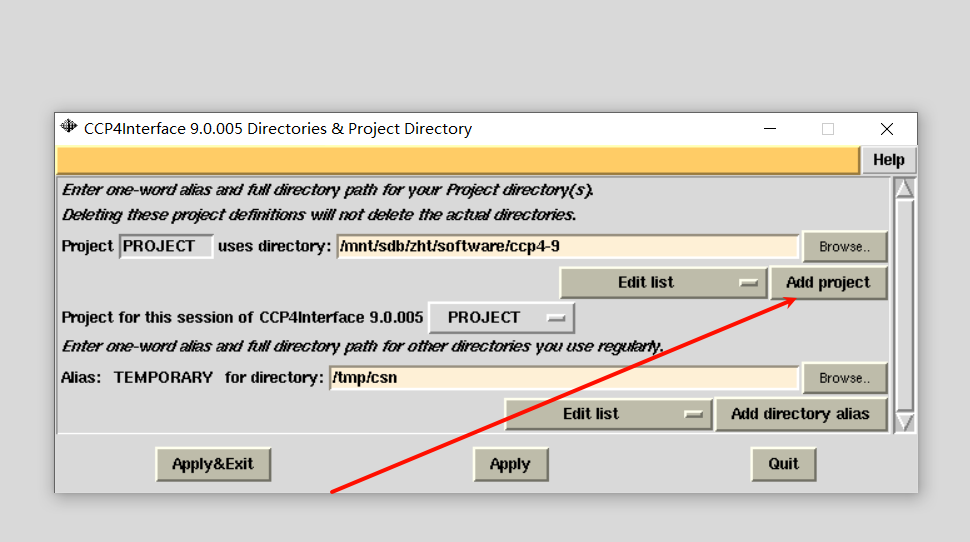
选择先前所存放的衍射数据文件夹:
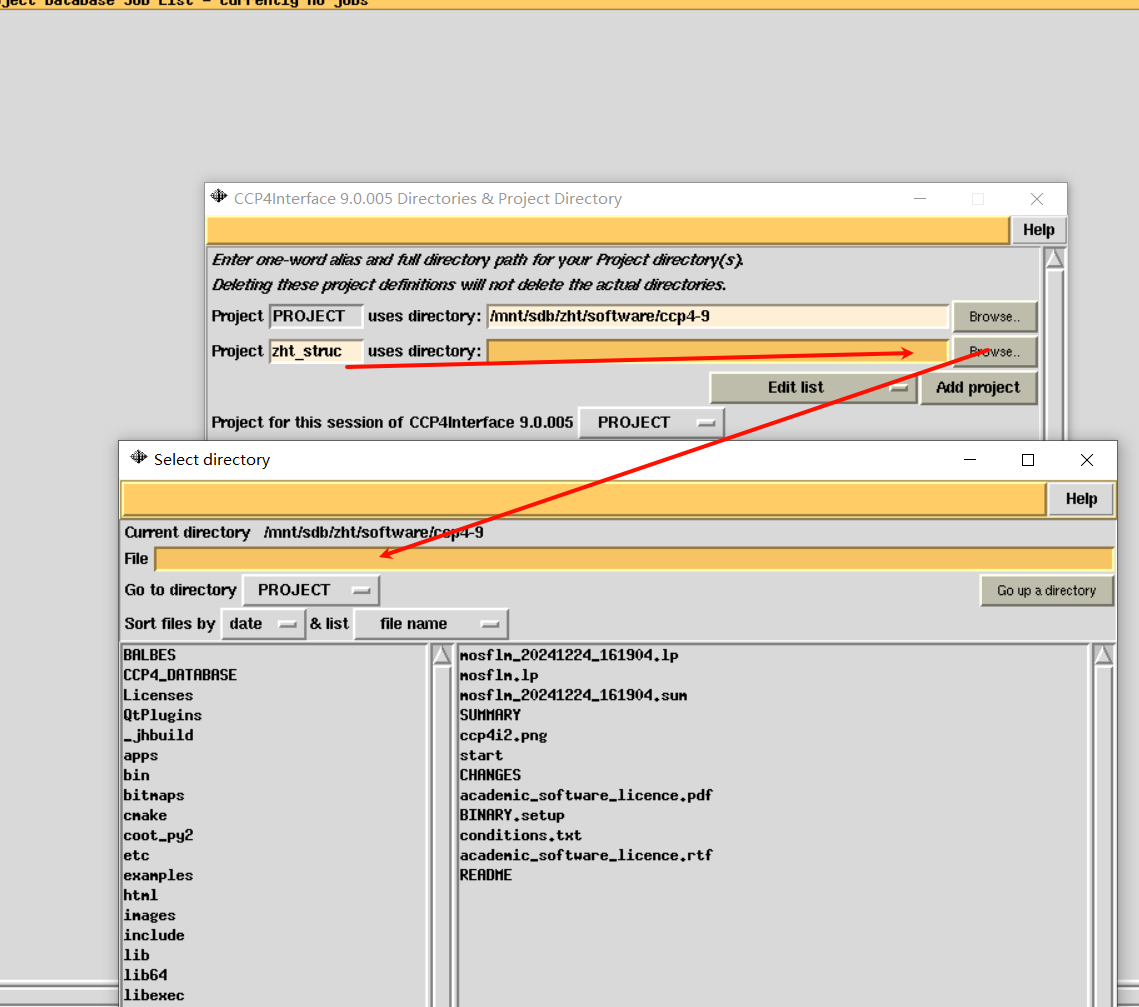
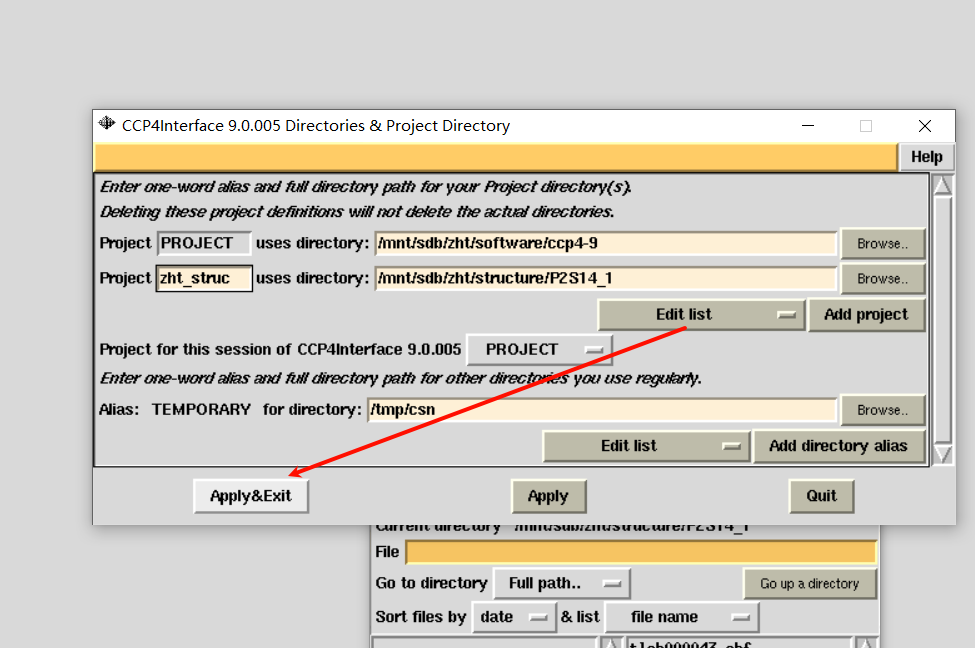
总之:
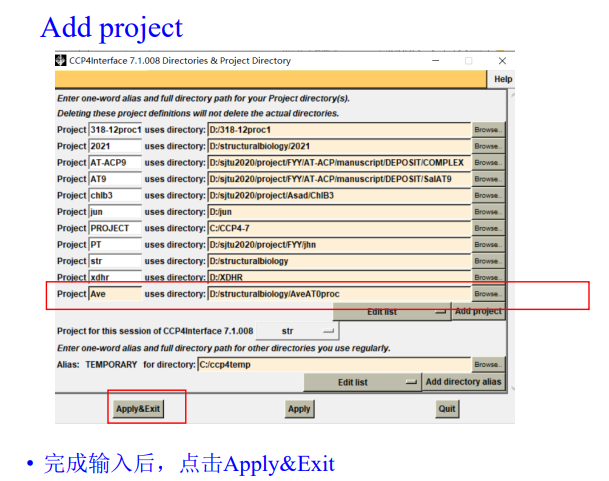
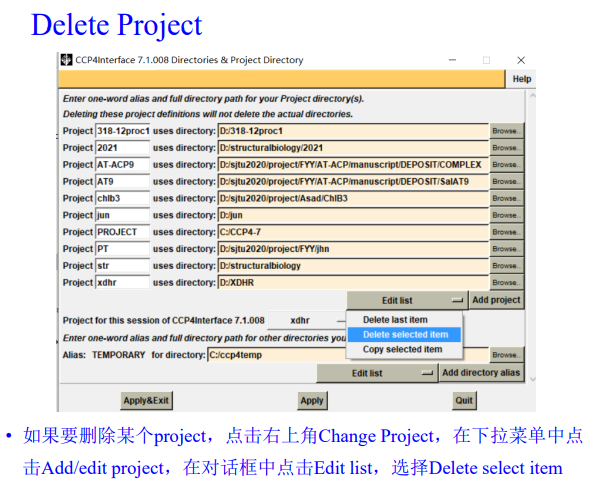
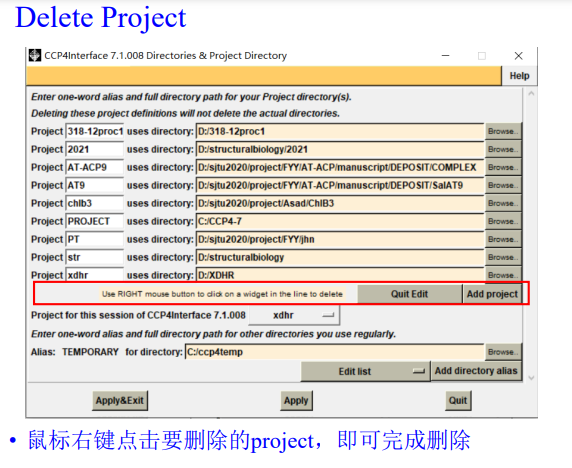
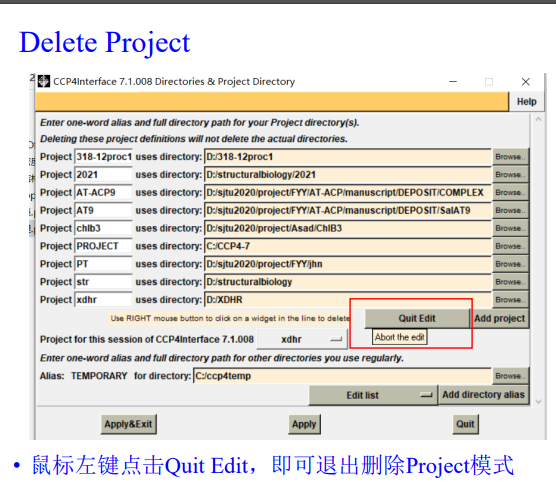
如果要删除项目:
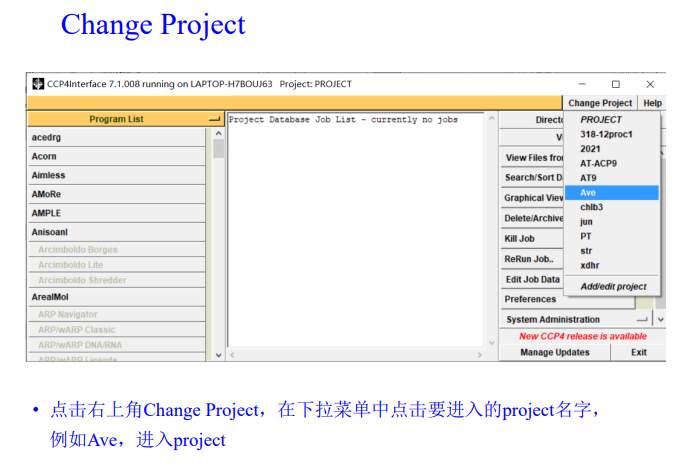
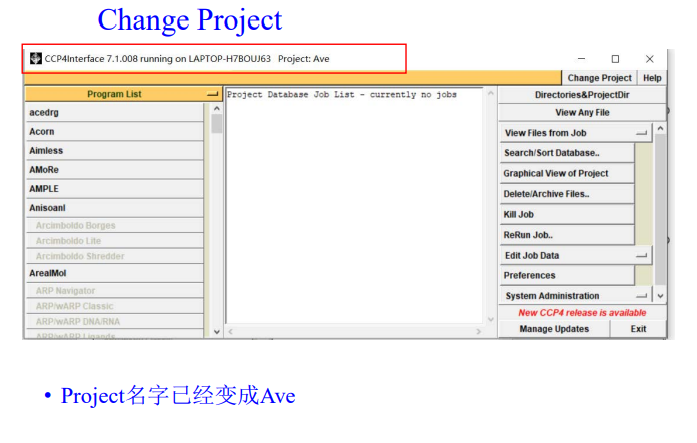
3,启用imosflm模块:在program list中
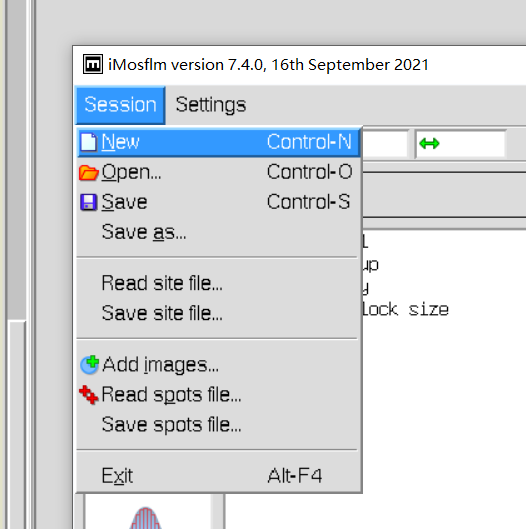
session:文件操作
settings:文件设置
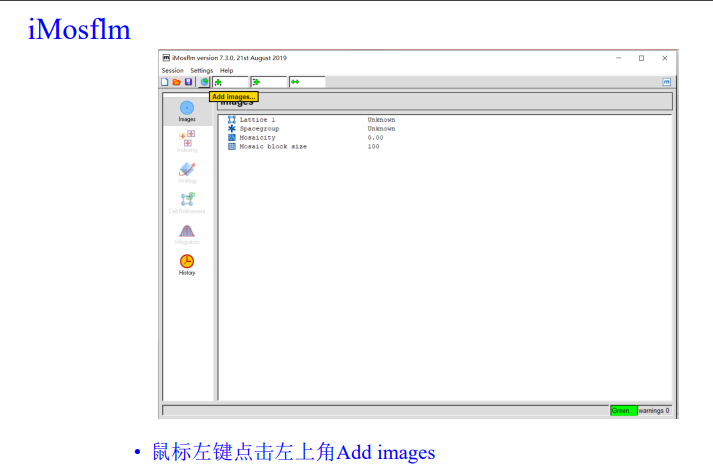
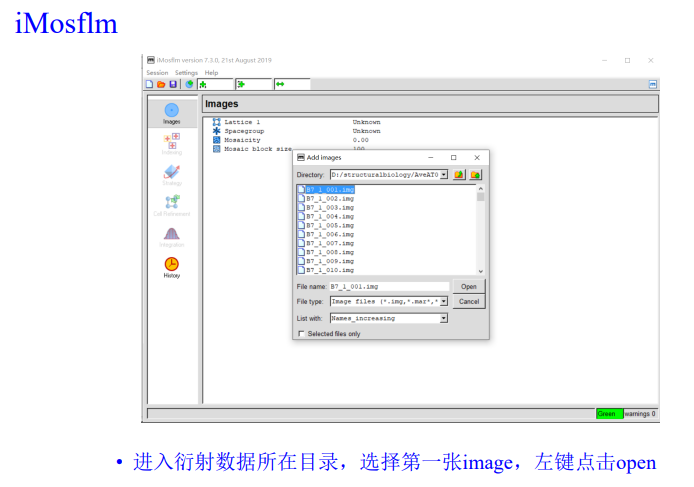
实操即
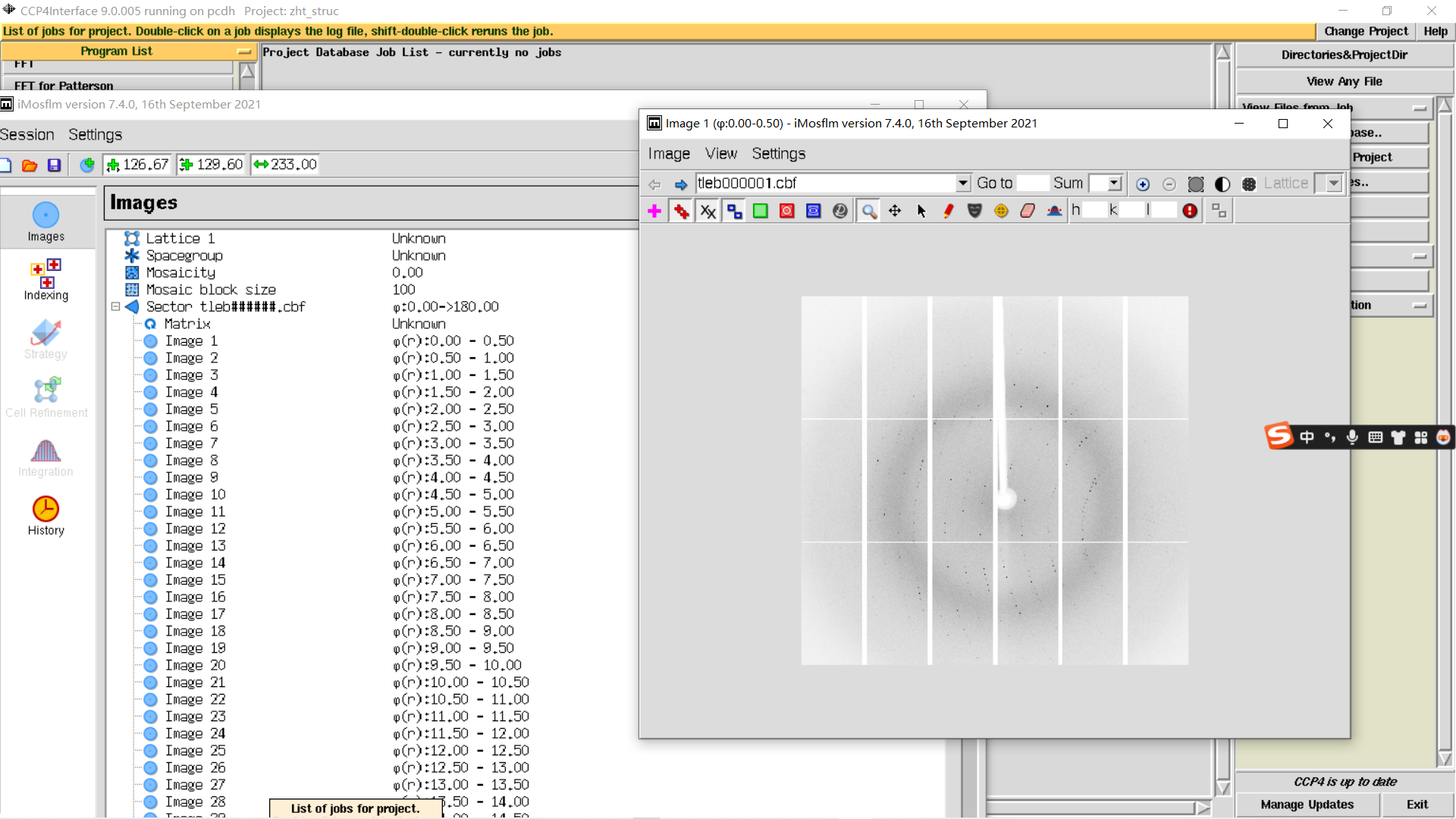
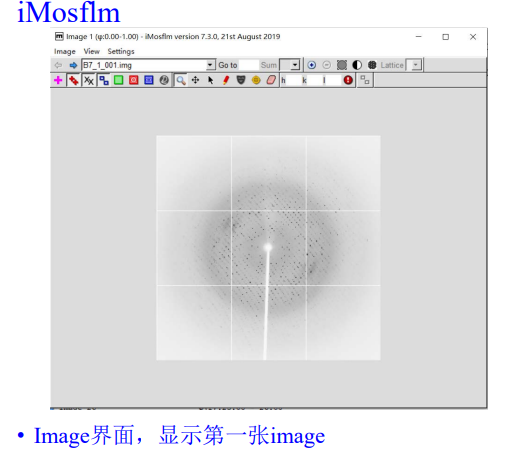
然后就是基础的图片文件操作:

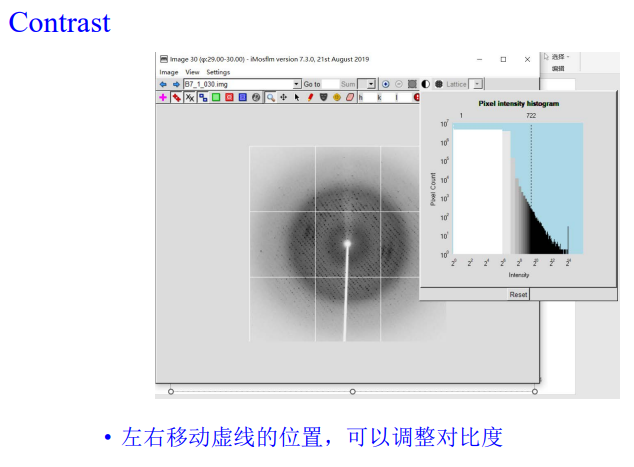
这里的对比度
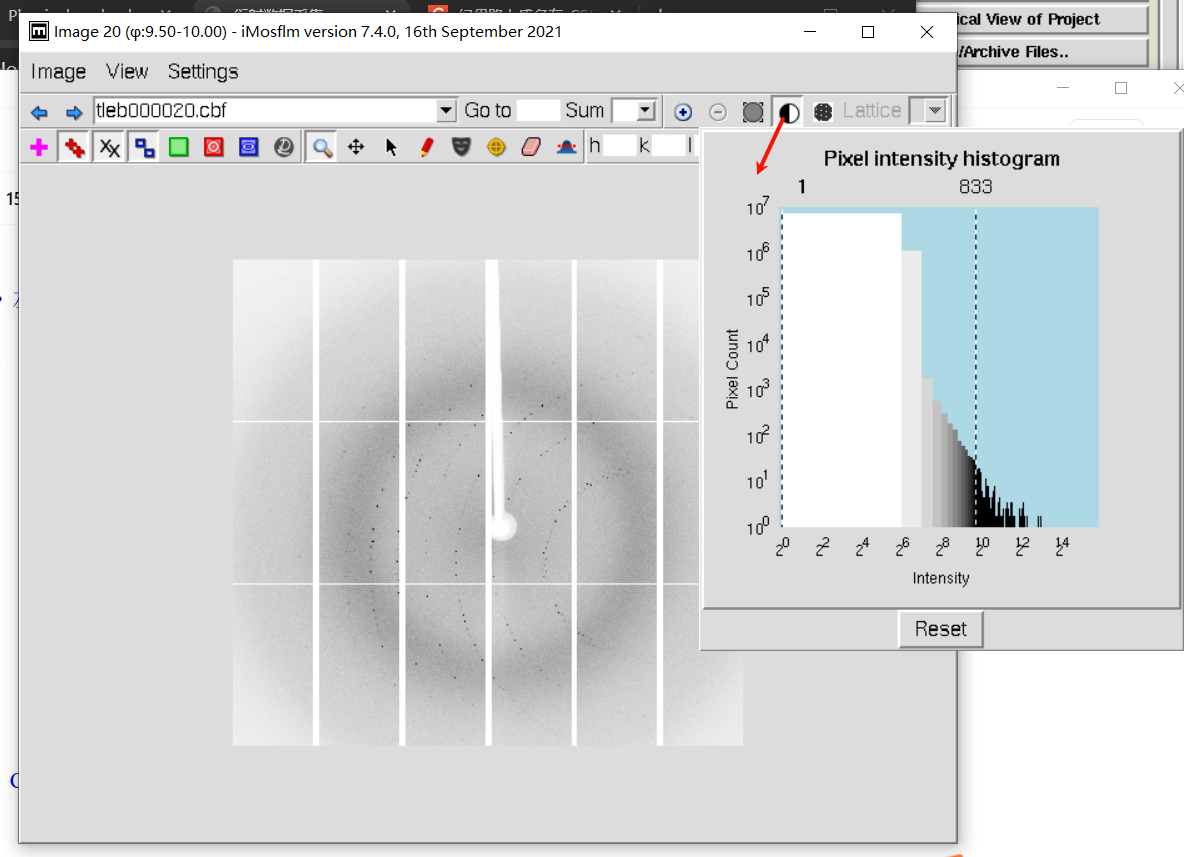
一般设置600-700左右
粉色的是中心
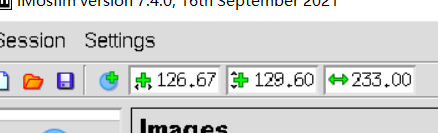
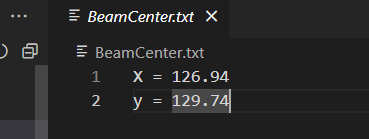
这个是写在文件里的

















 1413
1413

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









