






ANI(全基因组平均核苷酸身份)是衡量两个微生物基因组之间相似度的一个关键指标,它被定义为两个微生物基因组之间共享的直系同源基因对的平均核苷酸一致性。ANI广泛应用于微生物基因组学和系统生物学中,特别是在细菌或古菌的分类、基因组分类以及新物种鉴定方面。FastANI就是专门为快速计算这个指标而开发的,它基于短读序列比对技术,能够高效地评估不同基因组之间的相似性。与传统的全基因组比对方法相比,FastANI的速度更快,资源消耗更低,非常适合大规模数据的分析需求。
工作原理
FastANI将查询序列分割为短序列片段,然后使用基于MinHash的序列映射引擎Mashmap来计算同源映射并估计一致性。这种基于MinHash的方法不需要进行传统的序列比对,大大节省了计算资源,提高了计算速度。
功能特点
高速
FastANI的核心优势在于其无比对计算方法。传统的ANI计算需要进行耗时的序列比对,而FastANI通过使用Mashmap作为其基于MinHash的序列映射引擎,避免了昂贵的序列比对步骤。这种方法大大提高了计算速度,其速度比基于BLAST的方法快了2到3个数量级。
准确性
尽管FastANI采用非比对方法,但其准确性与基于BLAST的ANI求解器相当。如果ANI值远低于80%,则不报告基因组对的ANI输出值,建议在氨基酸水平上进行计算。
支持多种基因组类型
FastANI支持完整基因组组装体和草图基因组组装体的成对比较,适用于各种规模的基因组数据。这意味着无论你的基因组数据是完整的还是初步组装的,FastANI都能有效地进行ANI计算。这对于处理大量基因组数据尤其重要,因为它可以节省大量的计算时间和资源。
多种比较模式
FastANI提供了多种比较模式,包括一对一、一对多和多对多的基因组比较。这种灵活性使得它能够适应不同的研究需求,无论是比较两个基因组之间的相似性,还是批量比较多个基因组,FastANI都能轻松应对。
输出选项丰富
输出格式包括查询基因组、参考基因组、ANI值、双向片段映射计数和总查询片段数等详细信息。 用户可以选择生成phylip格式的lower triangular matrix文件,便于进一步的聚类分析。
可视化功能
FastANI还支持两个基因组之间计算的相互映射的可视化。通过使用--visualize标志,FastANI可以输出一个包含所有相互映射信息的映射文件,然后可以使用R脚本和genoPlotR包来绘制保守区域和热图。这种可视化功能可以帮助研究人员直观地理解基因组之间的保守区域和进化关系。
使用场景
FastANI广泛应用于微生物基因组比较、进化分析以及宏基因组研究等领域。例如,在研究细菌多样性时,你可以利用FastANI快速筛选出相似性较高的基因组,从而进一步进行深入分析。
总结
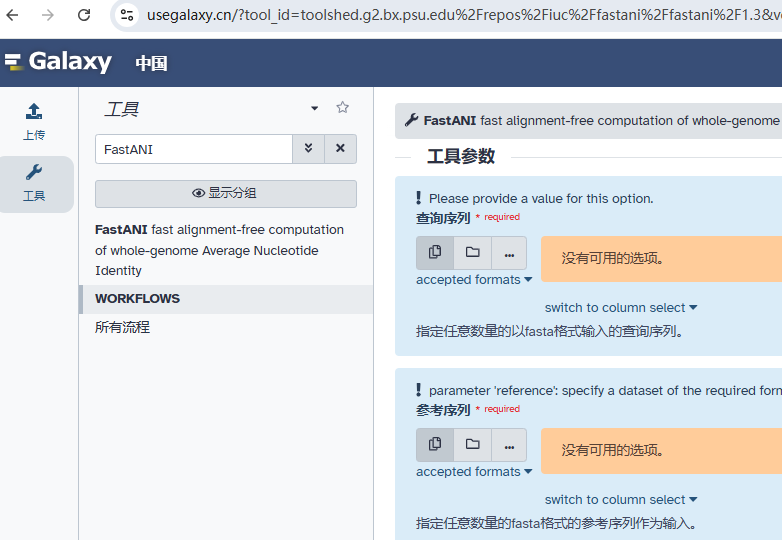
FastANI是一款非常优秀的生物信息学工具,在基因组的ANI计算方面有着显著的优势。FastANI依赖于Mashmap作为其序列映射引擎,这可能需要额外的安装和配置,对于不熟悉这些工具的用户,可以在Galaxy平台(usegalaxy.cn)上使用FastANI简单快捷地进行分析。希望这篇文章能帮助你更好地了解和使用FastANI,让你的研究工作更加高效。
推荐阅读
一键分析10X单细胞数据(点击图片跳转)
一键分析Bulk转录组数据(点击图片跳转)
简说基因 | 精选文章合辑(点击图片跳转)
生信平台
Galaxy生信云平台(UseGalaxy.cn)致力于降低生信分析门槛,让无专业背景的用户也能轻松分析数据。
• 界面化操作与强大的计算资源。
• 成百上千工具和流程免费使用。
• 丰富的可视化和交互分析工具。
• 强大的数据共享以及协作能力。
联系方式


















 2208
2208

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









