







题目:Virus classification for viral genomic fragments using PhaGCN2
发表杂志:Briefings in Bioinformatics
影响因子:13.994
发表时间:2022年12月03日
通讯作者:姜敬哲、原丽红、孙燕妮
第一作者:姜敬哲、袁文广、商家煜
通讯单位:
1. 中国水产科学研究院南海水产研究所
2. 广东药科大学
3. 香港城市大学
DOI:https://doi.org/10.1093/bib/bbac505
原文链接:https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbac505/6868523?redirectedFrom=fulltext
软件链接:https://github.com/KennthShang/PhaGCN2.0
- 摘要 -
病毒是地球上丰度最高、最多样化的生命体。鉴于病毒基因组的高度变异性,对其进行分类鉴定的难度较大,我们迫切需要一种高效而准确的病毒分类方法,尤其是对未知的新型病毒基因组序列进行快速鉴定。为此,我们提出了PhaGCN2—一个可以实现对整个病毒域(包括RNA病毒)进行快速分类和鉴定的工具。PhaGCN2不仅可以输出单条序列的科(Family)水平预测结果,还支持输出全部病毒序列分类集群网络图的可视化。我们从precision、recall、F1-score和需求的计算资源等方面将其与目前主流的病毒分类工具(vConTACT2、CAT和VPF-class)进行了对比,综合数据结果表明,PhaGCN2表现优于现有分类方法。基于PhaGCN2预测结果,我们将GOV2.0数据库中的可分类病毒序列数量提高了4倍,并且分类了90%以上的GPD数据库。PhaGCN2的使用将对病毒序列分类的标准化及国际病毒分类委员会数据库的快速扩展提供重要支撑。源代码可在以下网址免费获取https://github.com/KennthShang/PhaGCN2.0。
- 软件安装 -
只需要按如下步骤一行一行在Linux系统中输入,不报错即安装完成。
git clone https://github.com/KennthShang/PhaGCN2.0.git
cd PhaGCN2.0
conda env create -f environment.yaml -n phagcn2
conda activate phagcn2
export MKL_SERVICE_FORCE_INTEL=1
cd databasetar -zxvf ALL_protein.tar.gzcd ..
使用(ps:README中new function的版块是用来更新数据库的,如果您没有这方面的需求,请忽略。)
python run_Speed_up.py --contigs contigs.fa --len 8000
- 使用过程中至今收集到的问题 -
Q1:看不懂跑出来的结果,能不能麻烦指导一下哪些文件是可以用来做下一步的?
R:感谢您的提问,PhaGCN2文件中的final_prediction.csv是所有序列的预测结果汇总,其中第一列是序列号,第二列是索引号,第三列是我们给予的预测结果。
另外,为了节省在建图过程的内存使用,PhaGCN2是将大批量数据拆分成1000条/批次运行的,因此在network文件夹中输出的是各批次生成的网络图,其中的测试点例如0_1,对应的是pred文件夹中各批次的第二列的索引号,综上,如果您只是需要总的预测结果的话,您只需要final_prediction.csv即可,如果您需要网络图来对您的数据进行整体的可视化的话,您需要提取的是pred文件夹以及network文件夹。(ps:如果您需要在网络图中对点渲染颜色的话,在database/VMR_based_on_ICTV.csv 中存储着数据库的点对应的标准ICTV分类)
Q2:PhaGCN2支不支持设置输出路径?如果每次都只能生成在同一个目录下的话不太方便多次操作。
R:由于软件是分批次运行的,运行完后才可以得到一个总的结果,因此我们并不支持修改输出路径。当然如果您想同时运行多个程序的话,我们推荐您复制PhaGCN2的总的文件夹从而实现同时预测多个序列文件的操作。(另外,请注意如果同时运行多个程序时,可能存在内存不足的情况,请适当运行)
for example:
python3 PhaGCN2.0/run_Speed_up.py --contigs total1.fasta --len 2000
python3 PhaGCN2.1/run_Speed_up.py --contigs total2.fasta --len 2000
Q3:可不可以修改代码中的某些参数让程序包括1500-2000bp的序列?因为确实有些短序列也是来自于病毒。
R:目前PhaGCN2支持的最小病毒序列是1700bp,也即--len的参数需≥1700bp,(ps:由于目前已知最短的完整病毒基因组大概是1700bp)。虽然可以通过修改源代码的方式来支持预测长度更短的序列,但是一方面低于1700bp序列的precision我们没有测试过,另一方面低于1700bp的短序列不一定存在足够的可供分类的信息。我不建议您通过修改源代码的方式来预测1500-1700bp的序列(ps:如果要修改的话,会需要重新训练GCN模型,我们以后会考虑这个更新方向),您依旧可以通过使用PhaGCN2来预测您的序列中1700-2000bp的病毒序列。(ps:PhaGCN2在使用前会剔除掉不符合len参数以及含有存在未知gaps的序列)。
Q4:最近一次测试过程中,比如十万序列,拆开几份的分类结果和整个直接跑的结果居然有些contig分类会不一样,是什么原因呢?
R:在PhaGCN2的网络图中,存在一类自成聚类的family_like集群,此时该类集群中的family标签是由GCN算法由集群的综合特征给予的。在俩次不同的测试中,由于病毒簇的大小及簇中点的种类的差异,这时候就会有几率导致整个集群family_like的family标签出现差异。在俩次不同的测试中,虽然他们的结果不同,但这个聚类内部始终是联系紧密的。
Q5:这个like的真实性多高呢?比如family水平的like和它接近的这个科属于同一个纲的可能性很高吗?还是说最好当不能分类处理。
R:大多数基于学习的模型必须指定标签集(如科标签),这将无法容纳来自新科的病毒,为了解决这个问题,也为了更好的扩展数据库,我们在PhaGCN2中引入了family_like的概念,也即我们希望能通过like来发现一些具有明显规律性的病毒集群,即便这个集群在ICTV中从未见过(如文章中附图一的右上角放大图中的集群①-⑤)。一方面,在我们使用新增的2021年序列对PhaGCN2的测试中,他们之间同纲的可能性是非常高的。另一方面,在我们对测试序列与标准序列的建树中,大部分like的序列与其接近科的序列互成俩个不同的分支。综上,我们认为他们几乎是同纲的。另外,值得一提的是,为了节省在建图过程的内存使用,PhaGCN2是将大批量数据拆分成1000条/批次运行的,final_prediction.csv只是总结果,各批次分类的预测结果保存在pred文件夹中,网络图结果保存在network文件夹中。所以如果要藉此建树的话,我们推荐使用同一次的一千条序列进行建树。
Q6:最近做了个130000条contig的数据集,能分类的却有100000左右,是否偏多,还是说比较正常?
R:这种都是由数据库中的病毒序列类型决定的,目前的ICTV数据库中,噬菌体病毒的样本很多,因此如果测试序列中噬菌体偏多,能分类的病毒就会显著增加。例如文章结果部分对肠道菌数据库的分类中,能分类的病毒超过90%。
Q7:PhaGCN似乎将fasta文件中的每个contig都作为一个病毒基因组,并对fasta文件中的每个contig输出分类结果。然而当输入文件是一个viral bin时就会出现问题,比如说一个fasta文件中包含一个病毒基因组的几个contigs。删除viral bin中以“>”开头的行和换行符(\n)似乎是一个简单的解决方案,你认为这个方法合理吗?你有什么建议吗?
R:看起来你的问题可以看作是分节段病毒和不分节段病毒之间的问题。在我们对分节段病毒的测试中,结果表明,分节段病毒和不分节段病毒对准确性没有显著影响。但如果您确保这些contigs属于一个病毒,我们支持您合并这些病毒contigs。毕竟,基因组长度越长,它涵盖的信息就越多,预测就越准确。
网络图结果补充说明
尽管ICTV正努力提供持续更新的病毒分类系统,但现有的分类组别依旧不足以跟上多样化病毒快速积累的步伐。因此,PhaGCN2允许用户使用 "family_like"分类来发现可能的新分类群。具体来说,PhaGCN2应用GCN对图中所有节点进行预测。如果一个测试节点有一个预测的科标签 "A",并且不是任何训练节点的一阶邻居节点,它就会被定义为 "A_like",因此就产生family_like节点。仔细观察就会发现,这些节点中有许多聚类并不是大网络的一部分,而是自己自成一个簇(如补充图S1簇①-⑤)。这些簇往往代表了新的分类群——还没有包括在目前的分类系统中。当然也存在一些"family_like"节点与训练节点间接连接,如补充图S1中的簇⑥就是例子。总之,训练节点的二阶或二阶以上的邻居节点就会被预测为"family_like"类型节点。
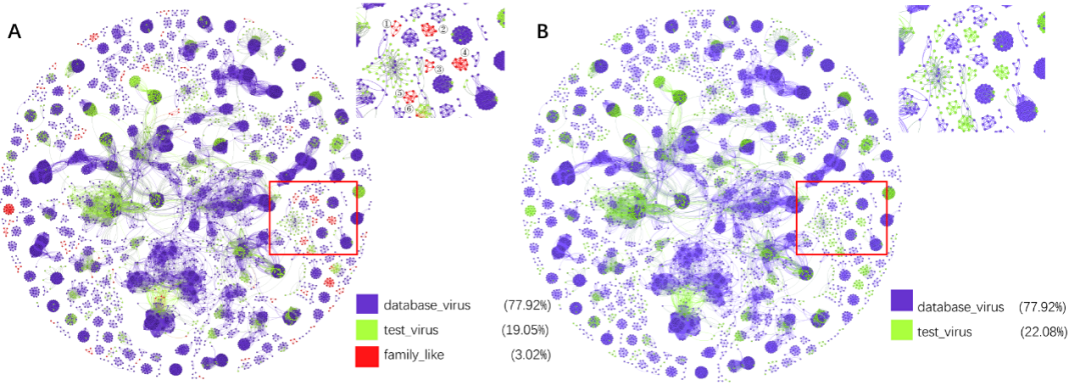
图S1. 应用 "family_like"之前和之后的网络图比较
A:PhaGCN2网络的可视化。B:PhaGCN网络的可视化。红框的放大部分在右上角。database_virus(紫点):ICTV 2020的参考基因组。test_virus(绿点)和family_like(红点)是测试数据(与ICTV2020相比,ICTV2021中新增加了2,636种病毒)。database_virus(紫点):训练节点。test_virus(绿点):与训练节点直接相连的测试节点。Family_like"(红点):与任何训练节点不相邻的测试节点。
- 结果 -
① PhaGCN2的改进处
PhaGCN2与前一版本相比有三个主要改进(补充表S1),包括(1)使用prodigal自动建立整个病毒域的蛋白参考数据库(补充表S2),(2)使用网络拓扑结构来帮助识别离群点,以及(3)将离群点分配给family_like。(2)和(3)的改进使PhaGCN2能够自动建议新的科,这消除了常用的监督学习模型中对固定标签集的限制。这些改进使PhaGCN2获得比原始版本更准确的预测,精度(precision)(公式(1))从73.19%提高到83.91%,灵敏度(recall)(公式(2))从87.92%提高到89.30%,F1分数(公式(3))从79.88%提高到86.52%(补充表S3)

② PhaGCN2与当前主流工具的比较
一致性(consistency):为了比较三种工具预测结果的一致性,我们以ICTV2021数据(9603个病毒基因组序列)作为测试数据。如图1A所示,vConTACT2和PhaGCN2都有预测结果的病毒数量为2248个,其中1494个相同,一致性为66.46%((739+755)/(1199+1049))(详细信息见表S5);PhaGCN2和CAT都有预测结果的病毒数量为6752个,其中5090个是相同的,一致性为75.39%((739+4351)/(1199+5553));vConTACT2和CAT都有预测结果的病毒数量为1266个,其中777个是相同的,一致性值为61.37%((739+38)/(1199+67))。三种工具都有预测结果的病毒数量为1199个序列,其中739个有相同的预测结果,一致性为61.63%(739/1199)。
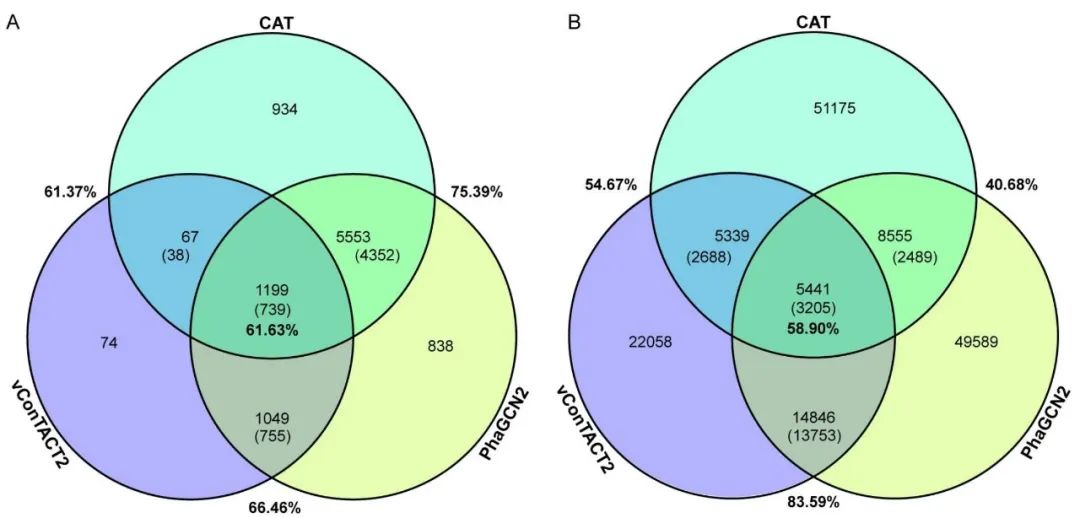
图1. 三种病毒分类软件结果的一致性
A:测试数据:9603条ICTV2021序列。B:测试数据:482,522条GOV2.0序列。不含括号的数字是有预测结果的数量,括号内的数字是相应工具有预测结果相同的数量,百分比是两个工具之间或三个工具之间的一致性。
我们进一步以GOV2.0(包括482,522个病毒基因组序列,其中大部分是未知病毒)作为测试数据。GOV2.0的文章中已经提供了vConTACT2预测的结果。因此,我们只运行PhaGCN2和CAT来预测GOV2.0(VPF-Class因其计算速度慢而并未纳入测试)。vConTACT2只获得47,839个预测结果(9.91%),CAT预测了170,200个病毒(35.27%),PhaGCN2获得199,833个预测结果(41.41%),高于CAT和vContact2。如图1B所示,vConTACT2和PhaGCN2都有预测结果的病毒数量为20287个,其中16958个病毒是相同的,一致性值为83.59%((3205+13753)/(5441+14846))(详细信息见表S6)。PhaGCN2和CAT都有预测结果的病毒数量为13996个,其中5694个相同,一致性值为40.68%((3205+2489)/(5441+8555))。vConTACT2和CAT都有预测结果的病毒数量为10780个,其中5893个是相同的,一致性值为54.67%((3205+2688)/(5441+5339))。三种工具共预测了5441个序列,有3205个序列的结果相同,一致性为58.90%(3205/5441)。上述结果表明,这些工具对已知病毒预测具有相似的一致性。但当关注未知病毒时,基于对齐的分类方法,如CAT,与其他工具的一致性较低。
准确率(Precision)、灵敏度(recall)、F1-score的比较:当使用ICTV2021中新增加的病毒作为测试数据时,PhaGCN2的灵敏度和准确率分别为89.30%和83.91%(表S1)。在此,我们进一步对PhaGCN2、vConTACT2、CAT和VPF-class用PhaGCN2数据库中收集的所有ICTV2021序列进行了测试,并将得到的预测结果与ICTV2021的分类进行了比较(表3)。如表3所示,PhaGCN2取其F1分数比第二好的工具CAT高10%;PhaGCN2的精度和灵敏度分别比CAT高7%和11%。结果表明,PhaGCN2在很大程度上比最先进的工具提高了病毒分类的精度和灵敏度。详细结果见表S5
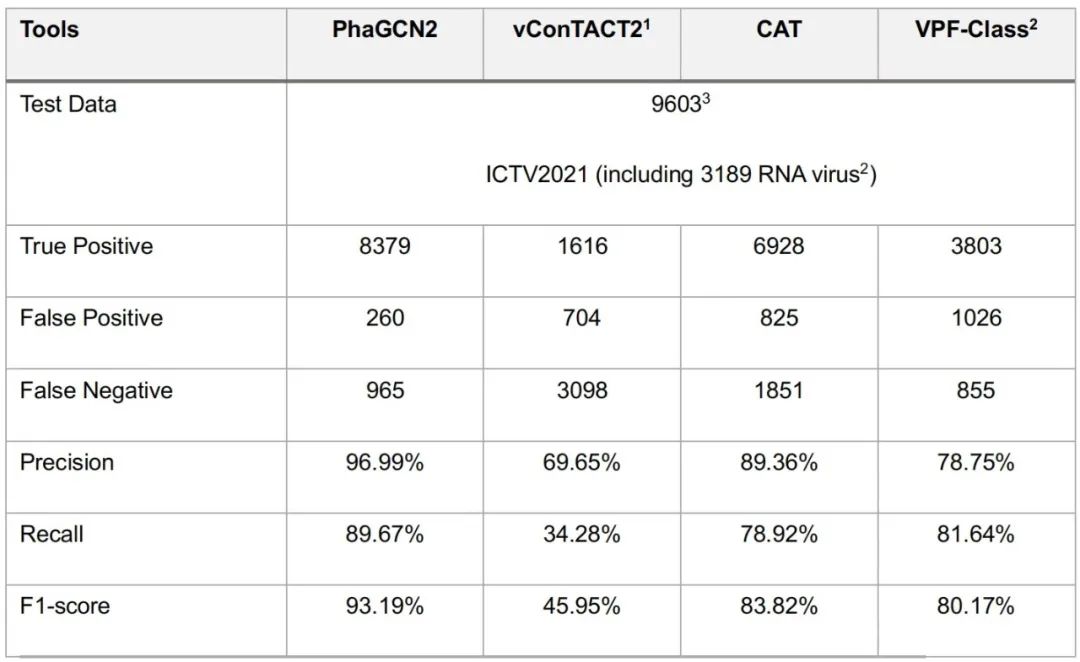
表3. PhaGCN2与最先进的病毒分类工具的比较
1 RNA病毒基因组被排除在vConTACT2测试数据评估之外,因为它仅用于DNA病毒分类。2由于VPF-Class测试数据仅用于DNA病毒和RT病毒分类,因此将Orthornavirae病毒基因组排除在VPF-Class测试数据评估之外。我们只将membership ratio和confidence score均高于0.2的结果作为VPF-Class的阳性结果。3以ICTV2021中大于1700 bp的病毒基因组作为测试数据,对所有软件进行评估。
③ PhaGCN2对属水平预测的可能性
和vConTACT2一样,PhaGCN2也可以绘制网络图。我们使用1700个人类肠道微生物组DNA病毒的基因组作为测试数据,并根据PhaGCN2的结果绘制网络图。我们渲染了数据库中最大的10个科的结果(图3A)。如图,同一科的病毒节点紧密地聚集在一起。为了在属水平上可视化簇,我们选取了病毒种最多的Siphoviridae。同样,在图3B中,Siphoviridae的前16个属成员用不同的颜色显示出来。我们可以看到一些属,如Pahexavirus,Skunavirus和Ceduovirus,自身形成非常紧密聚类簇。然而,一些属(如Triavirus、Phietavirus、Bioseptimavirus、Dubowvirus和Peeveelvirus)则是混在一起(图3B)。这表明它们的差异不足以使PhaGCN2预测它们是不同的属,另一方面,也提示了ICTV的现行分类标准还有进一步的提升空间。
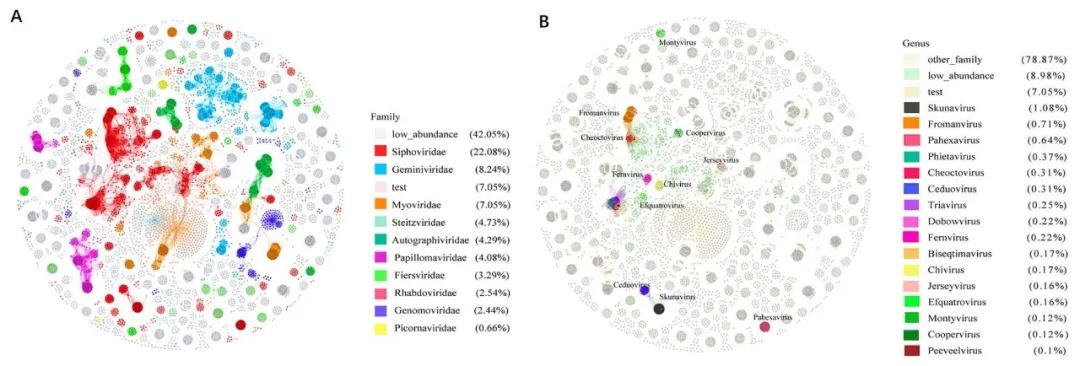
图3. PhaGCN2网络图在科和属水平上的聚类效果展示
AB两图的拓扑结构完全相同,test为MGV测试数据。A:图中丰度为前9的科用不同颜色标识,没有着色的low_abundance代表其它低丰度的科;B:图中专门显示Siphoviridae科内的不同属,高丰度属(成员数量≧10)用不同颜色标识,low_abundance用浅绿色标识,代表Siphoviridae中其它低丰度的属,other_family代表非Siphoviridae科。
④ PhaGCN2对公共数据库的挖掘
GPD和GOV2.0代表了两个完全不同的病毒生境。在本文中,我们使用PhaGCN2对GPD和GOV2.0数据库进行分类。去除不合格序列后,它们分别剩下142333和328173。如图4所示,GPD和GOV2.0的总体预测成功率分别为91.9%和40.8%。GOV2.0中未知病毒的比例较高,远远超过GPD,说明海洋中的病毒还没有得到充分表征,是病毒多样性研究的重要资源库。仅从分类类别来看(不包括unknown), Siphoviridae和Myoviridae占GPD的54.5%,Siphoviridae_like和Myoviridae_like占GPD的31.1%。与GPD相比,在GOV2.0中Siphoviridae和Myoviridae占28.9%,Siphoviridae_like和Myoviridae_like占40.4%。如果包括Caudovirales下的其他科,如Podoviridae和Herelleviridae,人类肠道中99.16%的噬菌体为Caudovirales,而海洋中为94.8%。这意味着在目水平上,GPD和GOV2.0以Caudovirales为主,而在科水平上GPD和GOV2.0差异较大。具体结果见表S9和表S5。
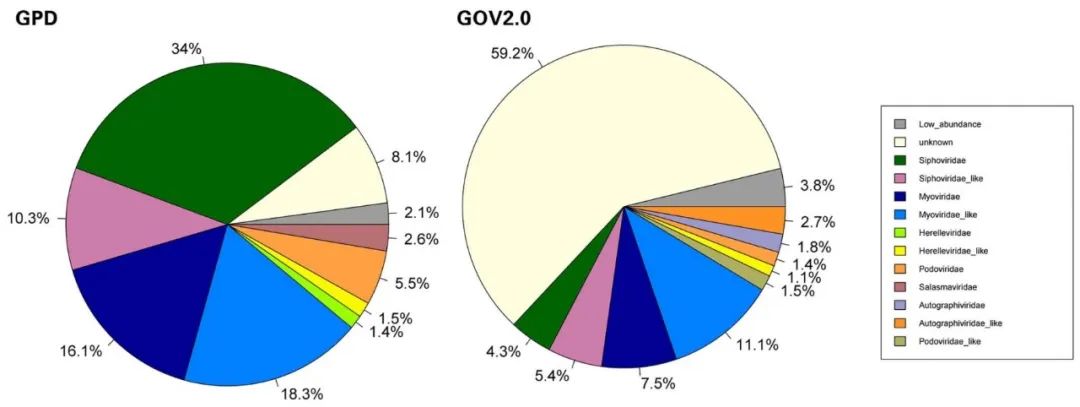
图4. 基于PhaGCN2的GPD和GOV2数据中的病毒科水平组成结果比较
该图为GPD和GOV2数据库中使用PhaGCN2进行分类后根据各个科占据的百分比画出来的饼状图,其中“-“后面的数字代表序列条数,low_abundance表示数量较少(低于总个数的0.5%)的科的总和,unknown代表未能预测的数量,其他分别代表各个科。GPD和GOV2.0总测试点分别为142809和482522。
进一步,针对GOV2在科水平的分类及来源站位信息,我们还绘制了Myoviridae和Siphoviridae在不同站点和海水深度的分布丰度图(图5),详细经纬度及其含量数据见附表11。由图5可见,Myoviridae表现出非常明显的纬度和深度效应,越接近赤道地区、表层水域,Myoviridae占比越高,而Siphoviridae则是俩极区域显然要超过赤道地区。这说明不同科的病毒可能在漫长的进化过程中对同环境进化出了独特的适应性。
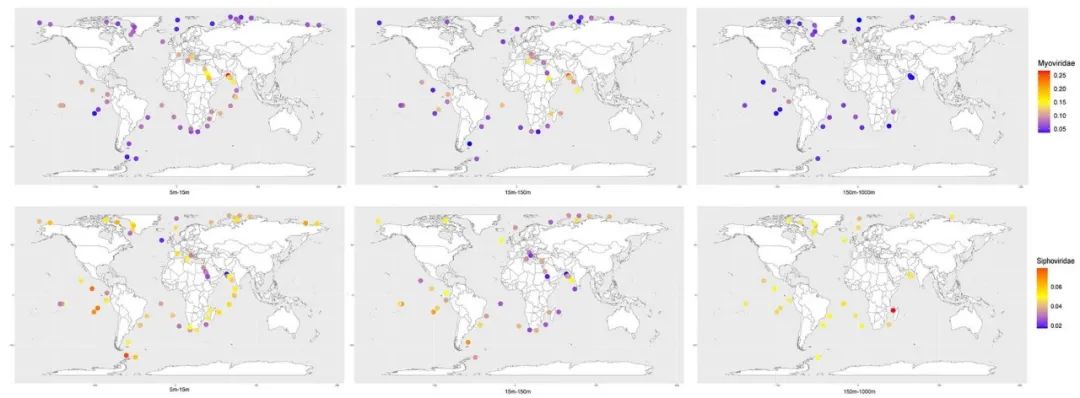
图5. GOV2的Myoviridae和Siphoviridae病毒类群随纬度和海水深度的不同分布模式
地图中颜色深度代表Myoviridae和Siphoviridae占该取样点中总病毒种类百分比含量的大小,颜色代表含量的高低。从左至右的采样深度依次为5m-15m,15m-150m,150m-1000m。
- 讨论 -
与CAT、vConTACT2和VPF-Class相比,PhaGCN2的一个局限是不能进行科以下的分类。虽然理论上我们的方法可以扩展到属水平,但许多属的成员数量较少,不足以训练一个广义的学习模型。消除这一限制是我们未来的工作。一个理想的属级分类工具应该解决一些额外的挑战。首先,属的数量明显大于科。目前,根据ICTV的报告,大约有2200个属。此外,各属的基因组数量形成了高度不平衡的分布,给稀有属的分类也带来了挑战。第三,目前ICTV标准下的一些属过于相似,无法有效区分(图3B)。由于我们的工作重点是科级分类,所以在属级上介绍当前工具的详细比较超出了本工作的范围。随着ICTV参考数据集的不断增长和ICTV对近亲属的调整,在基因水平的预测将更加可行。
与其他基于学习的模型一样,PhaGCN2的性能也依赖于训练数据的质量。由于测序的偏差,目前的训练数据并没有系统地覆盖全部的不同的分类组。尽管PhaGCN2利用网络拓扑结构来建议新的科,但其对新科的预测能力依旧是有限的。同源性低于37.4%的未知病毒序列的检测率通常很低(图S2)。加强新病毒科分类的一个可能策略是使用PhaGCN2进行迭代预测。首先,我们可以用PhaGCN2对所有病毒基因组数据(如IMG/VR)进行预测。然后,我们可以将新预测的Family_like成员加入训练数据,以提高PhaGCN2识别更多新科成员的能力。迭代训练和搜索可能会提高PhaGCN2对新科的检测能力。我们将在今后的工作中对此进行研究。
- 通讯作者 -

中国水产科学研究院
姜敬哲
研究员
博士生导师
姜敬哲,博士,研究员,中国水产科学研究院“贝类病害与生态防控创新团队”首席专家(PI),广东省环境功能基因芯片工程技术研究中心主任,广东省“特支计划”科技创新青年拔尖人才(2014),研究方向包括:(1)创新、升级病毒(组)学研究方法和工具,发现、鉴定新型海洋病毒和潜在疫病病原;(2)运用微生物生态学、分子诊断和培养组学等研究理论和方法,进行病害预警、养殖健康调控、噬菌体制剂等方面的应用开发。主持包括国家自然科学基金等各类项目总经费超过1000万元,在Microbiome、Plos Pathogens、Briefs in Bioinformatics、Aquaculture等期刊发表学术论文50余篇,获国家发明专利授权十余项,成果转化2项,获得海南省科技进步一等奖1项、广东省科技进步二等奖1项。

广东药科大学
原丽红
教授
研究生导师
原丽红,博士,广东药科大学教授、研究生导师,生物工程一级学科负责人,海洋药学专业负责人,广东省生物技术候选药物研究重点实验室学术带头人。主要从事“生物活性物质体外合成与利用;多组学技术解析生物功能基因进化、环境适应和传播扩散机制”相关研究。先后前往伦敦大学和布利斯托尔大学(2008)、中国科学院南海海洋研究所(2010-2012)、悉尼大学(2013-2014)、渥太华大学(2016)等国内外知名高校和科研机构进行长期访问和交流,取得了一系列重要的理论突破和原创性成果。获得广东省自然科学二等奖1项(排名第一,2018)、广东省科技进步二等奖1项(排名第六,2017)、广州市珠江科技新星(2012)、中国科学院王宽诚博士后工作奖(2010)等科技奖励和荣誉称号;主持国家自然科学基金(面上、青年)、国家重点研发计划(子项目)、省市等各级科研项目18项,做为主要完成人参与国家、省、市科研项目10余项;发表学术论文60余篇,研究成果被Nature Reviews Genetics, PNAS, Genome Biology等Top期刊引用和评论;做为第一发明人获得授权发明专利9件(中国发明专利6件、美国发明专利2件、日本发明专利1件)、软件著作权2件;培养博士和硕士研究生15人。担任国家林业科技专家、广东省政府职能转移专家、广东留学人员联谊会-广东欧美同学会第二届理事会常务理事、广东省动物学会第十四届理事会理事等职务为社会服务。

香港城市大学电机工程系
孙燕妮
副教授
博士生导师
孙燕妮,香港城市大学电机工程系(Electrical Engineering)副教授,博士生导师。在美国圣路易斯的华盛顿大学(Washington University in Saint Louis)取得计算机系博士学位后在密西根州立大学计算机系担任助理教授和副教授(with tenure)。2018年加入香港城市大学。主要研究方向是生物信息学,序列分析,宏基因组学,和病毒基因组学。具体的研究课题,发表的论文,以及实验室的位置请参加作者个人主页:https://yannisun.github.io/。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 3319
3319

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









