






点击蓝字 关注我们
iMeta 高被引论文合集
iMeta 自2022年2月发行以来共发表110篇文章,引用超过10次的文章有16篇,其中Sangerbox、Majorbio Cloud和imageGP等工具平台引用均超100次。

Sangerbox: 交互式整合临床生信分析平台
https://onlinelibrary.wiley.com/doi/10.1002/imt2.36
Sangerbox (http://vip.sangerbox.com)是基于网络的工具平台,用户可以在一个友好的交互页面中进行不同的分析。平台提供可交互的图形化分析工具,包括相关性分析工具,通路富集分析、WGCNA分析等常见的工具和功能。
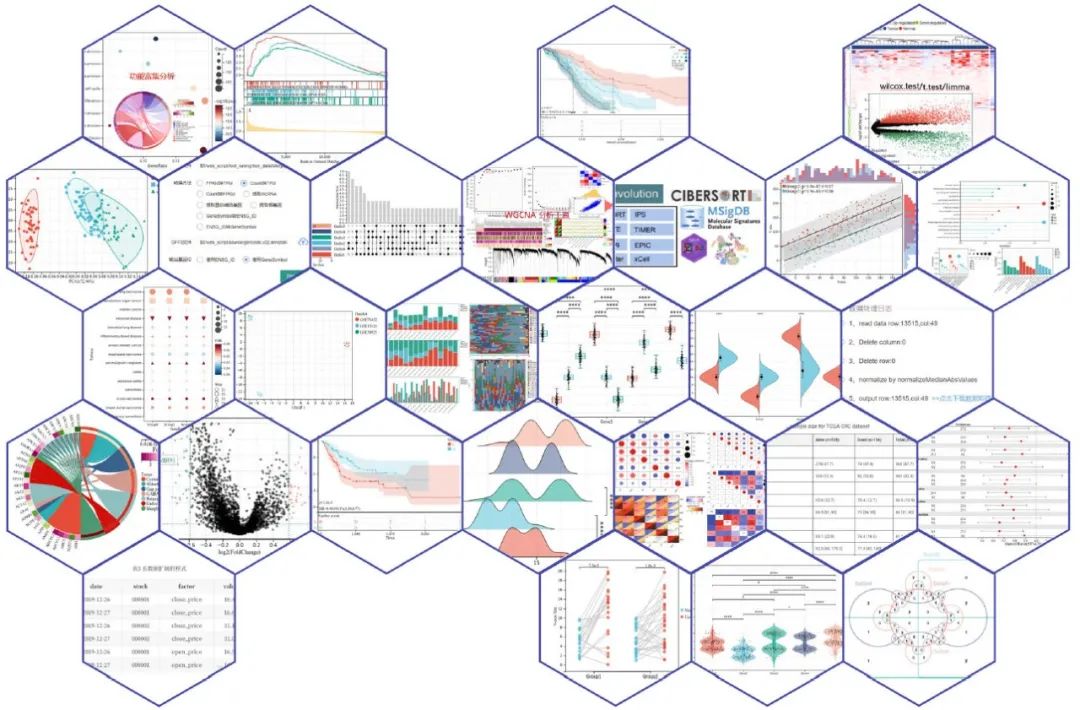
► 点击阅读
下载:6824;被引:186

Majorbio Cloud:一站式多组学数据分析平台
https://onlinelibrary.wiley.com/doi/10.1002/imt2.12
Majorbio Cloud是一站式高通量组学研究平台(https:// cloud.majorbio.com/),包括云流程、云工具、云课堂3大功能模块。其中云流程涵盖了微生态、转录组、蛋白与代谢及基因组等多组学,通过强大、快捷的分析流程,助力用户自主完成数据分析和挖掘。

► 点击阅读
下载:14616;被引:122

imageGP: 高颜值高被引绘图网站
https://onlinelibrary.wiley.com/doi/10.1002/imt2.5
ImageGP(http://www.ehbio.com/ImageGP/)是一款简单易用、功能强大的在线绘图工具 , 可以在线生成常见的线图、柱状图、散点图、箱线图、集合图、热图和直方图等。同时 ImageGP 还支持火山图、富集分析泡泡图、主成分分析类图等 和 4 种微生物领域的功能预测和生物标志物鉴定分析,包括 FAPROTAX, BugBase, PICRUSt 和 LEFSe。
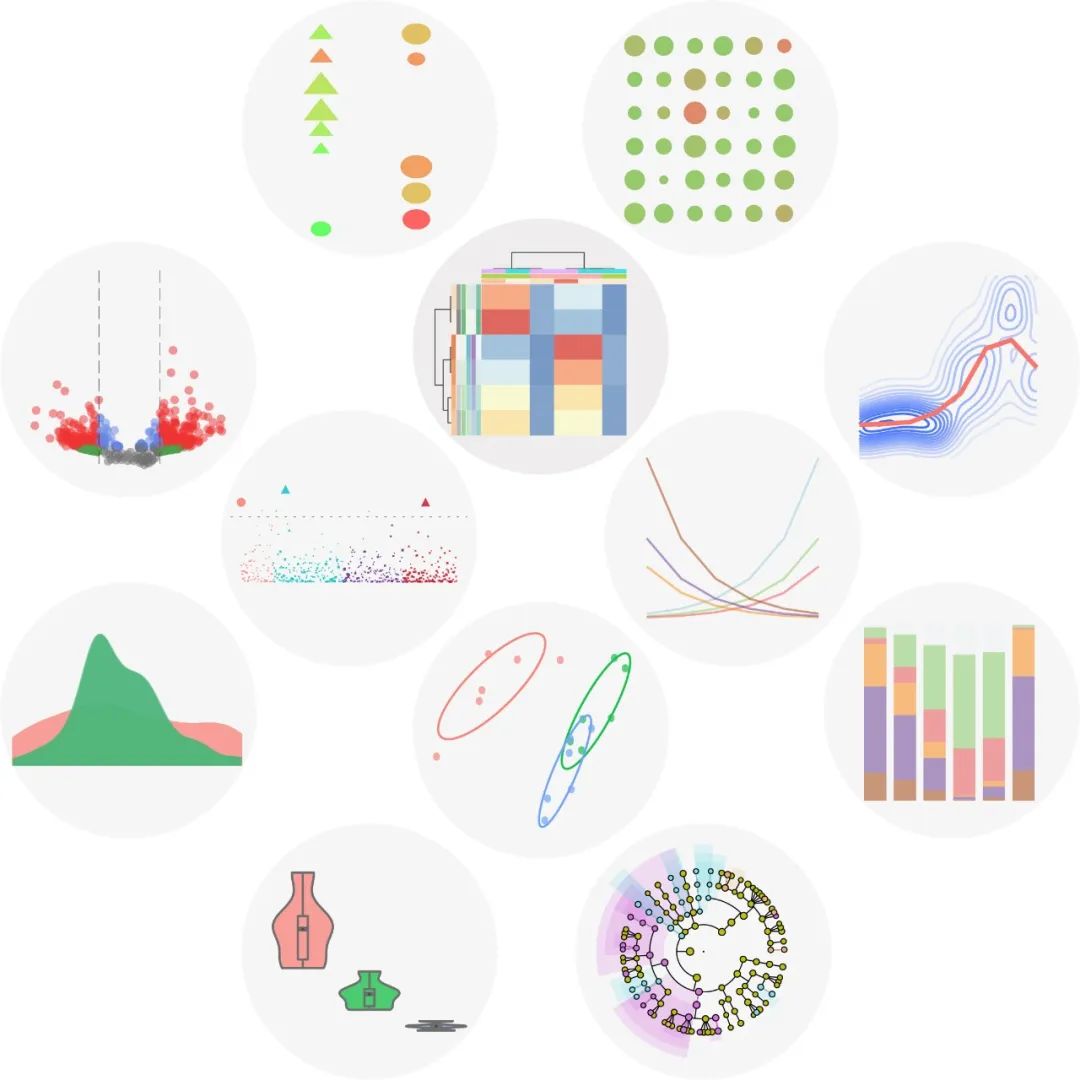
► 点击阅读
下载:12789;被引:119

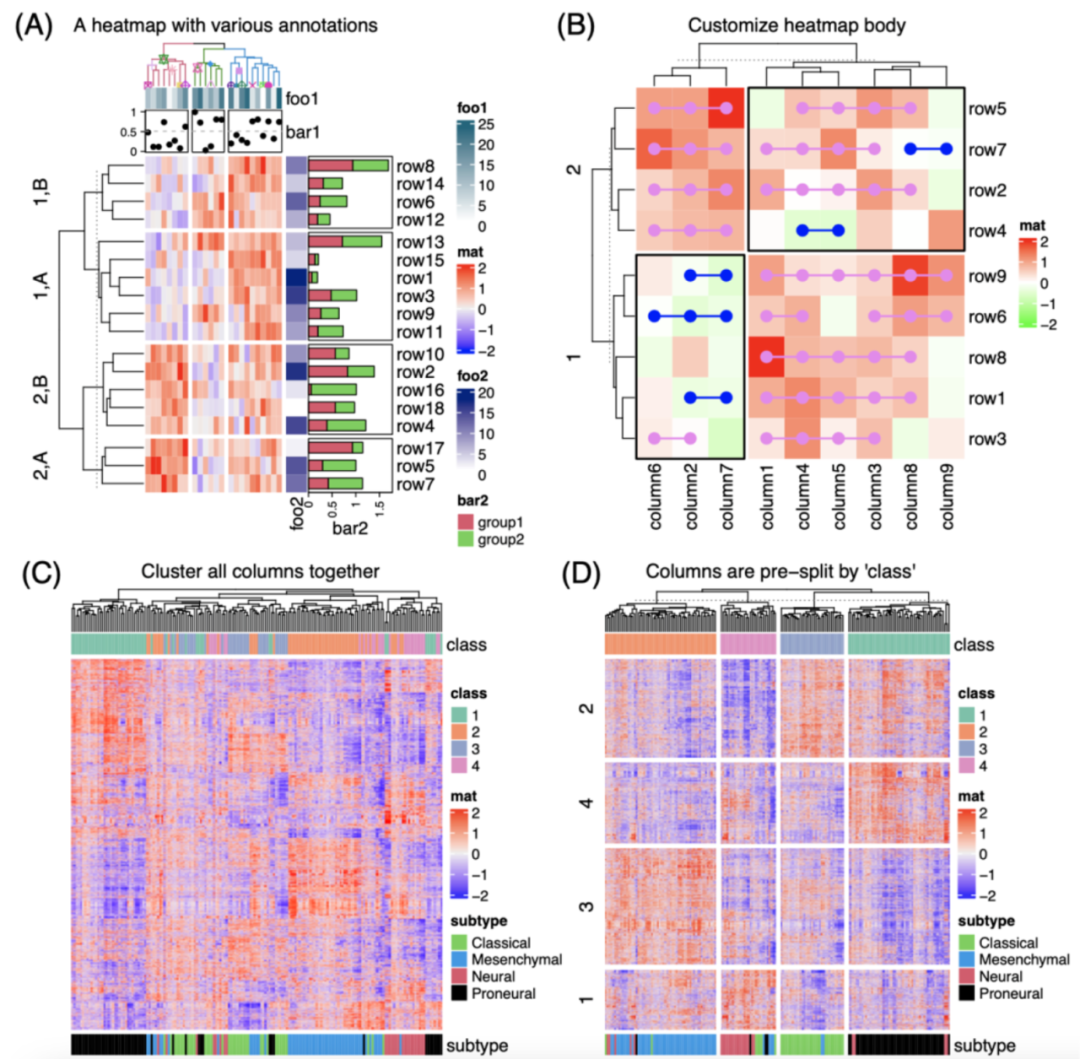
Complex heatmap: 复杂热图可视化
https://onlinelibrary.wiley.com/doi/10.1002/imt2.43
该研究系统性地介绍了 ComplexHeatmap R包在复杂热图可视化方面的特性和功能。ComplexHeatmap 可以通过自动拼接和调整多个热图以及添加复杂注释,轻松建立多个来源信息之间的关联,因此 ComplexHeatmap 被广泛应用于许多领域的数据分析中,特别是生物信息学,以发现隐藏在数据中的关键结构。
稳定版本发布在:
https://bioconductor.org/packages/ComplexHeatmap/
开发者版本发布在:
https://github.com/jokergoo/ComplexHeatmap
文档发布在:
https://jokergoo.github.io/ComplexHeatmap-reference/book/
论文中绘制图1到图6的代码发布在:
https://github.com/jokergoo/ComplexHeatmap_v2_paper_code

► 点击阅读
下载:17467;被引:73

iNAP: 针对微生物组学研究的集成网络分析平台
https://onlinelibrary.wiley.com/doi/10.1002/imt2.13
iNAP针对不同分类学水平的微生物物种,提供了物种域内关联和跨域间物种关联的网络分析流程,即分子生态网络分析平台(MENAP)和跨域生态网络分析平台(IDENAP)。为便于微生物组学数据的生态网络分析,iNAP提供了SparCC、eLSA、SPIEC-EASI和基于RMT理论的Pearson/Spearman相关性等构建关联的方法。iNAP平台免费注册,操作简单,无需生物信息分析和编程知识,即可快速方便的完成网络分析。
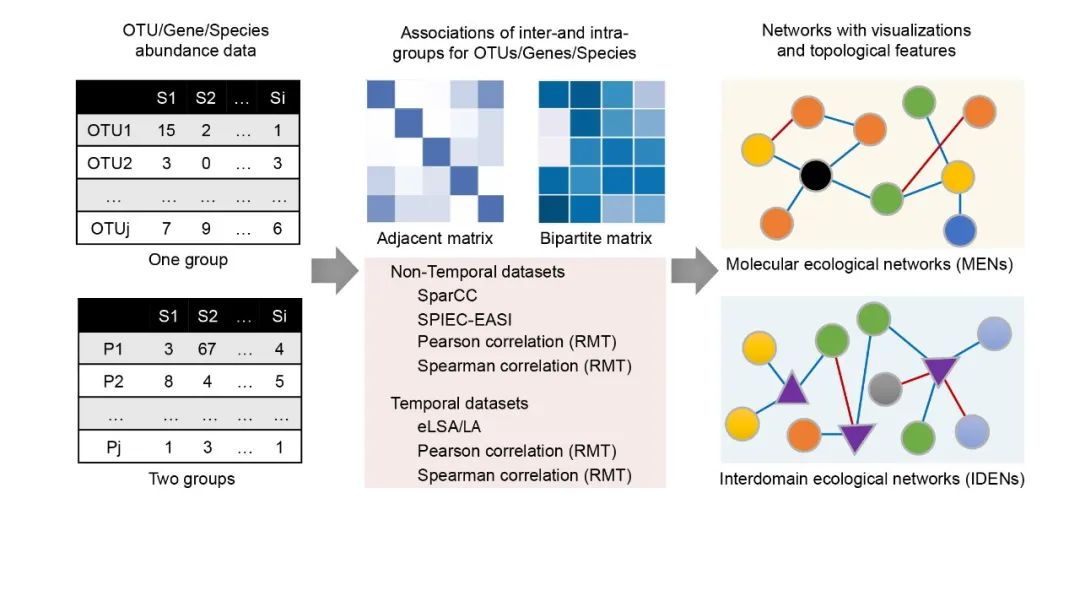
► 点击阅读
下载:6974;被引:64

干旱生态系统中土壤真菌与细菌群落构建的关系
https://onlinelibrary.wiley.com/doi/10.1002/imt2.2
本研究将跨生物群落的观测研究与微宇宙实验相结合,研究西北旱区复杂陆地生态系统中生物因素(如土壤真菌和跨界物种的相关关系)对土壤细菌生物地理和群落构建的影响。结果表明,土壤中真菌丰富度调节了细菌群落构建过程的平衡,随机过程随着真菌丰富度的增加而减少。
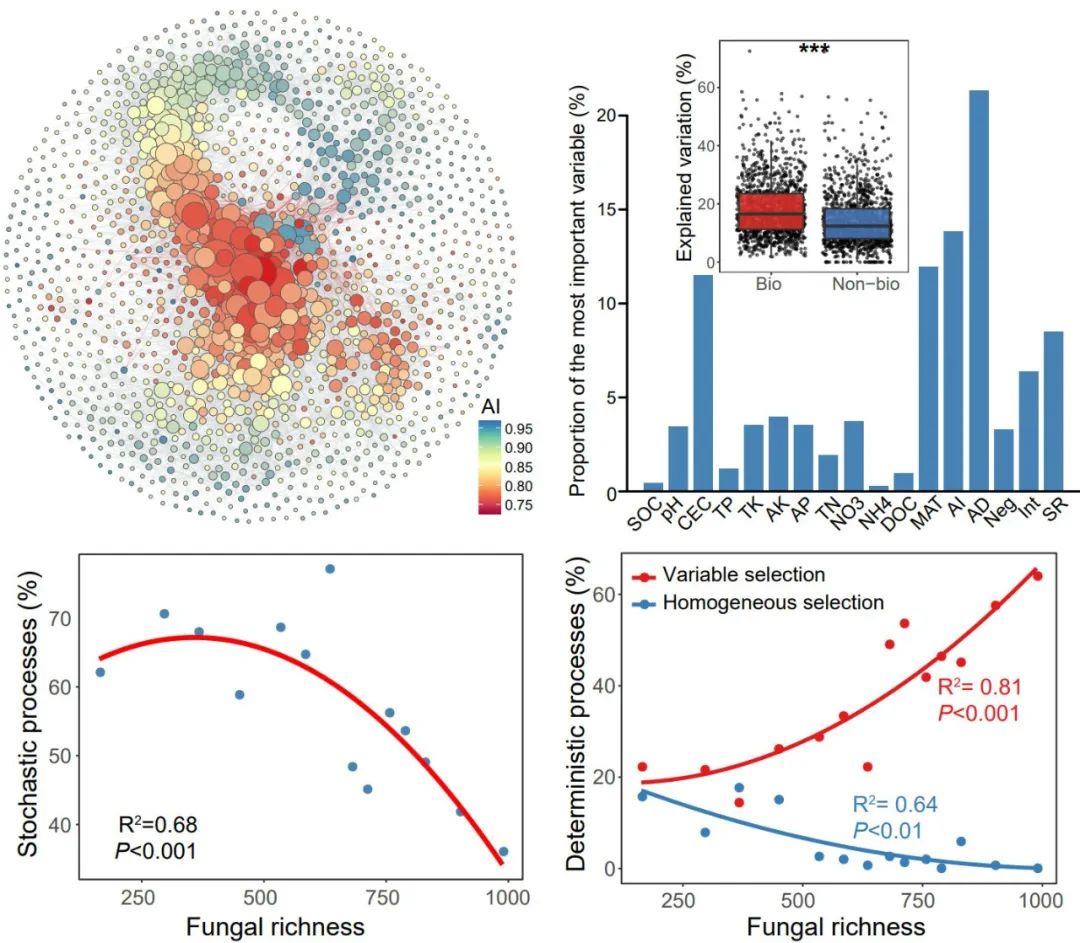
► 点击阅读
下载:7932;被引:38

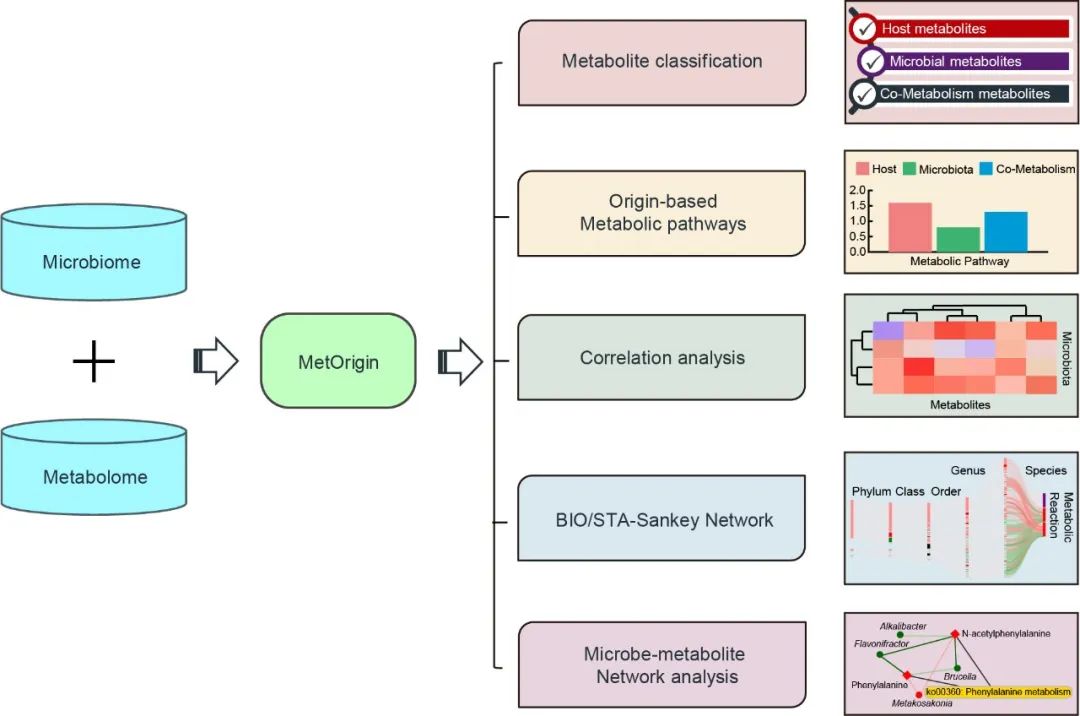
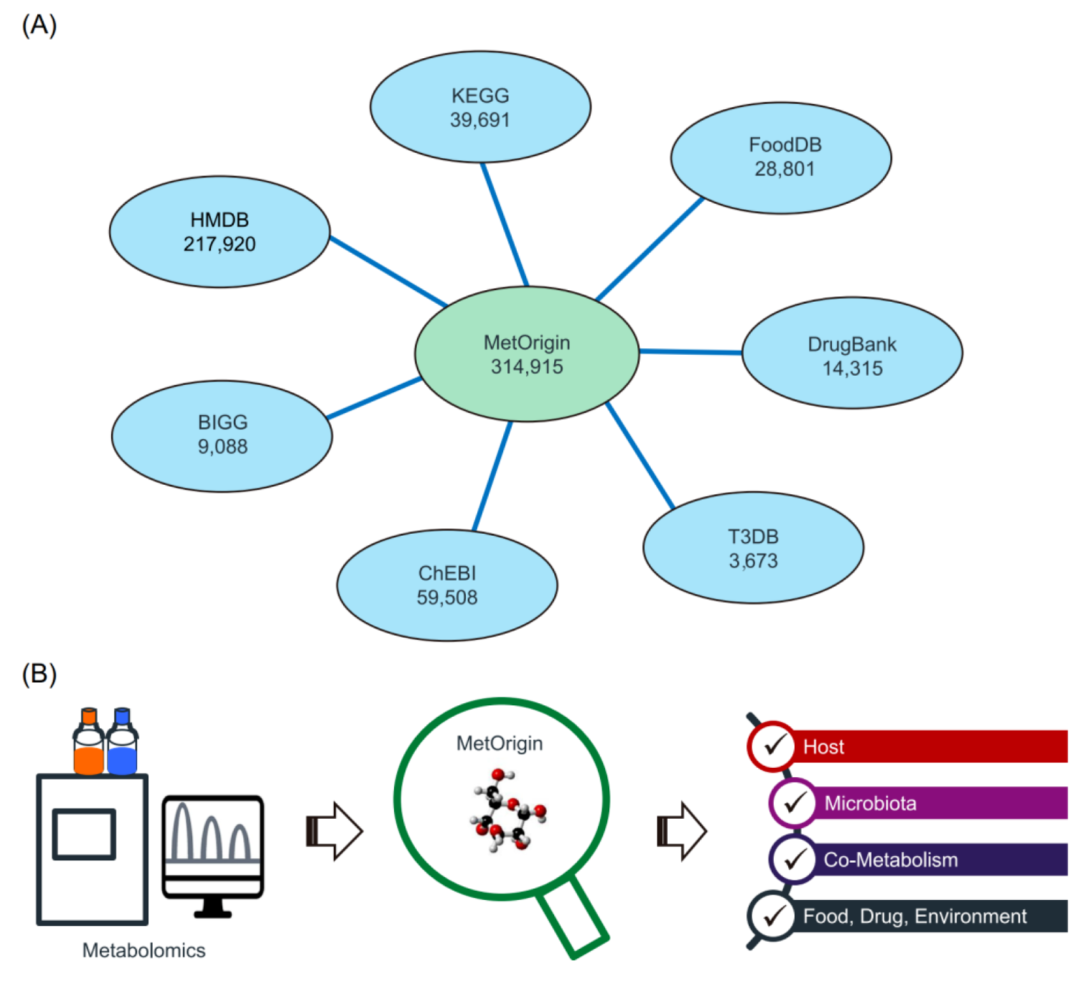
MetOrigin:代谢物溯源推动肠道微生物和代谢整合分析
https://onlinelibrary.wiley.com/doi/10.1002/imt2.10
MetOrigin(http://metorigin.met-bioinformatics.cn/)能够对代谢产物进行溯源分析。不仅可以快速识别微生物来源代谢物及其代谢功能,还有助于发现与其密切相关的关键微生物。
► 点击阅读
下载:5538;被引:31

ggClusterNet:包含多种基于模块可视化布局算法的微生物网络挖掘R包
https://onlinelibrary.wiley.com/doi/10.1002/imt2.32
ggClusterNet的R包,用于更加容易的进行网络数据分析挖掘和可视化。在ggClusterNet包中设计了数十种网络布局算法用于更好的展示微生物网络模块化信息(randomClusterG, PolygonClus-terG, PolygonRrClusterG, ArtifCluster, randSNEClusterG, PolygonModsquar-eG, PolyRdmNotdCirG, model_Gephi.2, model_igraph, and model_maptree)。
ggClusterNet在github(https://github.com/taowenmicro/ggClusterNet/)和Gitee(https://gitee.com/wentaomicro/ggClusterNet)上开放使用

► 点击阅读
下载:8091;被引:27

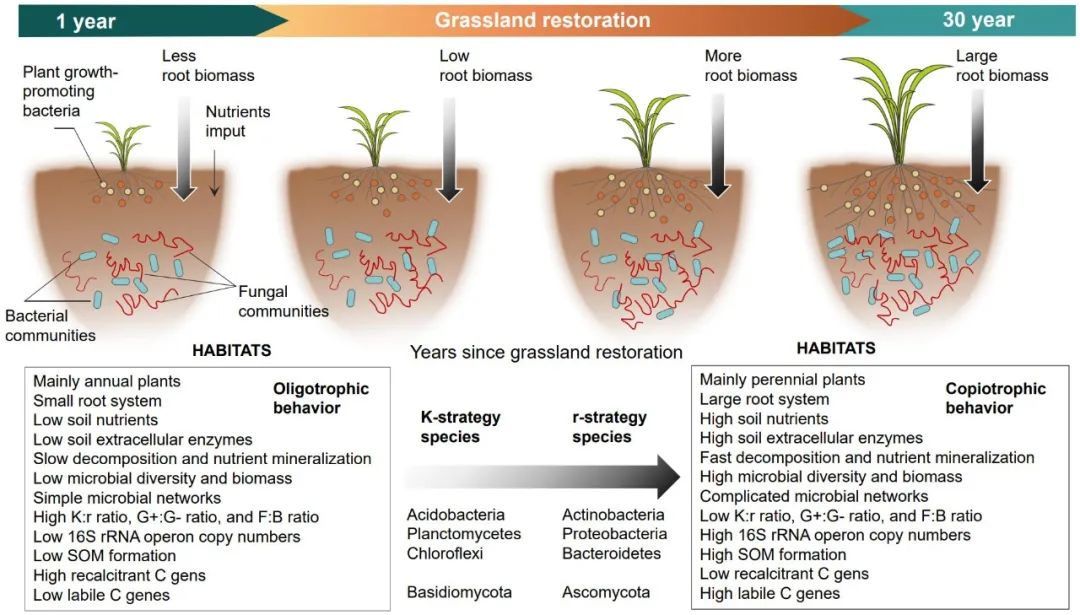
解译恢复草地土壤微生物生活史策略及其影响因素
https://onlinelibrary.wiley.com/doi/10.1002/imt2.66
本研究构建了一个框架,以突出草地恢复过程中植物和土壤特性在驱动微生物生活史特征方面的重要性,表明微生物生活史特征支持rRNA操纵子拷贝数能够反映土壤微生物资源有效性这一观点。
► 点击阅读
下载:2056;被引:19

Ggtree:用于系统发育树及相关数据存储与可视化的数据结构
https://onlinelibrary.wiley.com/doi/10.1002/imt2.56
该文章设计了ggtree对象用于存储系统发育树,相关数据以及可视化指令,提高了系统发育数据的可重复性与可重用性。
Ggtree软件包可以在
https://www.bioconductor.org/packages/ggtree/
免费获得,在线书籍
https://yulab-smu.top/treedata-book/
也提供了ggtree的完整参考。

► 点击阅读
下载:4923;被引:18

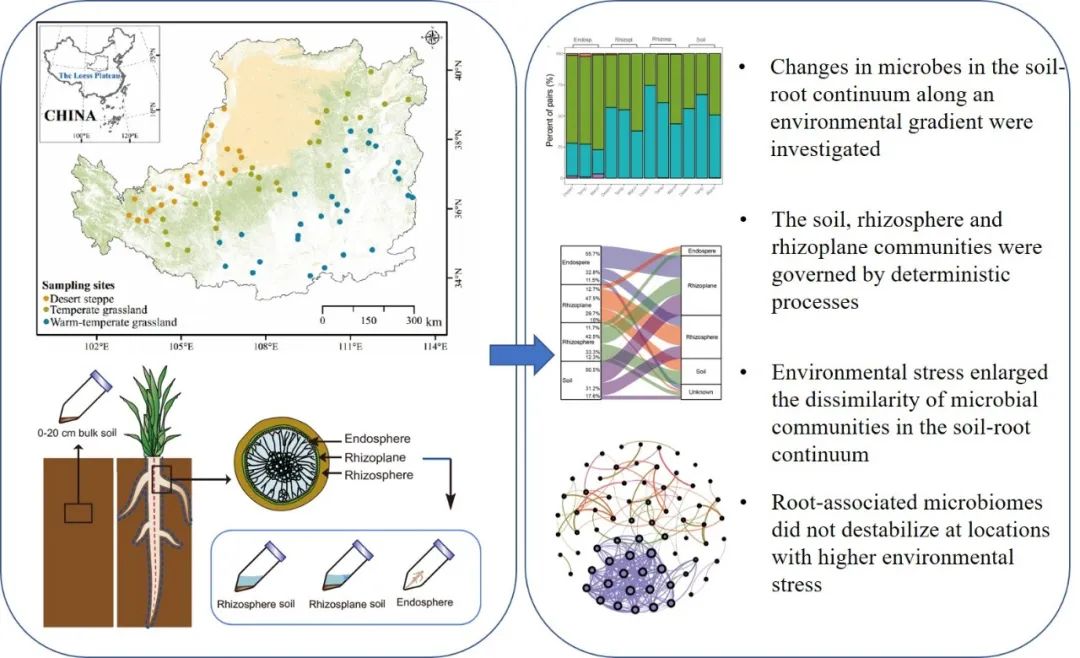
根系菌群沿环境梯度的响应机制
https://onlinelibrary.wiley.com/doi/10.1002/imt2.18
该研究表明微生物生态位依然是区域尺度上影响细菌多样性和群落组成的最重要因素,土壤-根系连续体中的细菌群落沿着环境梯度表现出不同的敏感性和组装机制,这受到了根系生态位与环境因子的共同影响。
► 点击阅读
下载:3515;被引:17

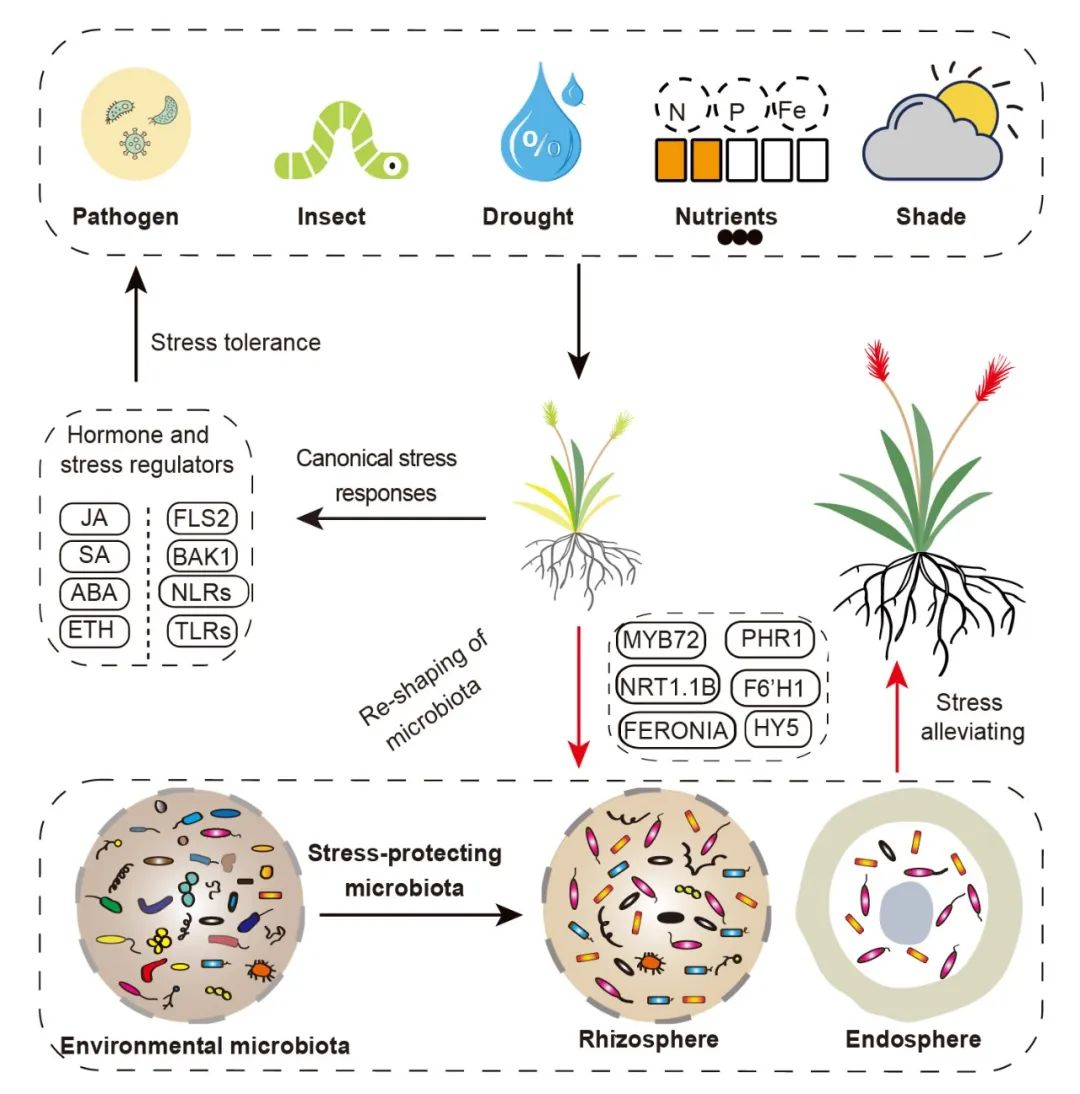
植物向微生物群“呼救”策略的遗传基础
https://onlinelibrary.wiley.com/doi/10.1002/imt2.8
该文章系统地回顾了宿主主动重塑有益微生物组,从而增强生物和非生物胁迫耐受性的遗传基础,总结了确立微生物组变化与植物功能性状联系的实用研究手段。
► 点击阅读
下载:3623;被引:16

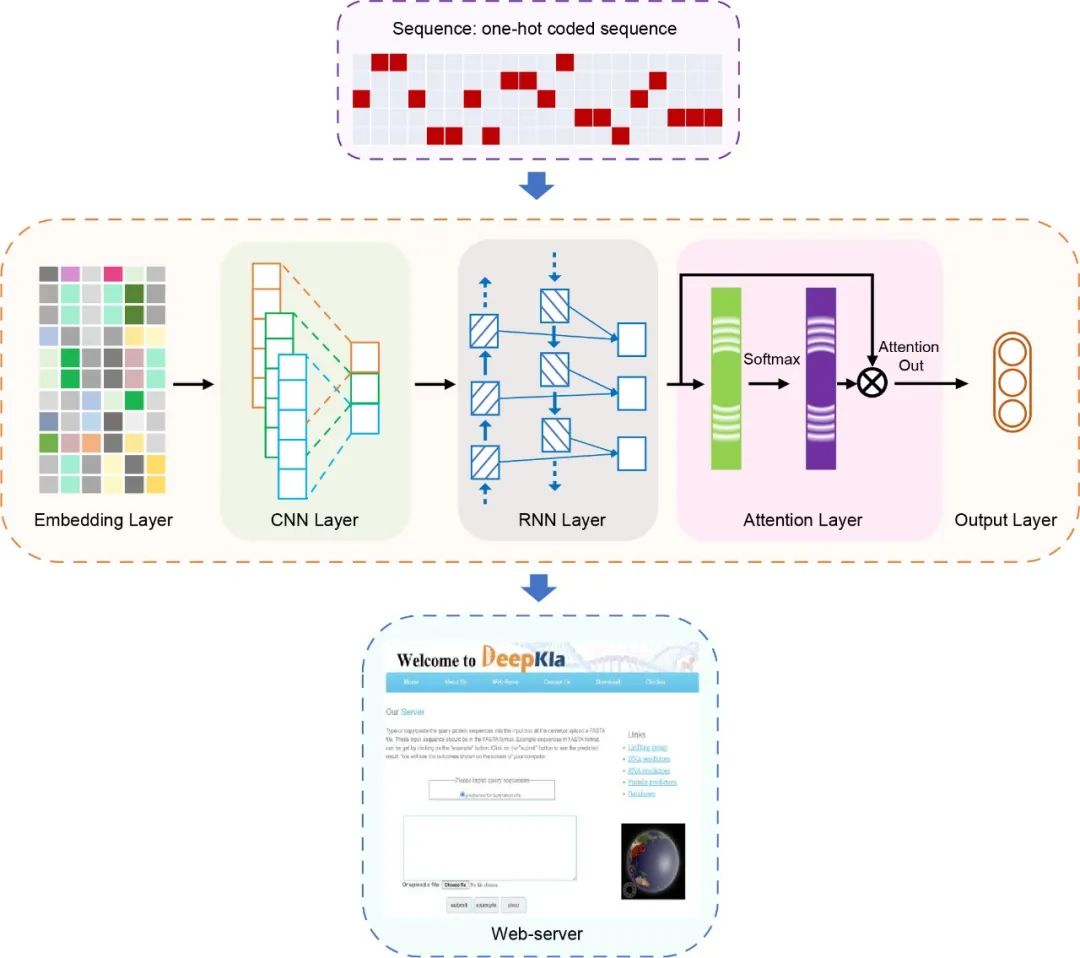
DeepKla:基于注意力机制的蛋白质赖氨酸乳酸化位点预测工具
https://onlinelibrary.wiley.com/doi/10.1002/imt2.11
该研究提出第一个用于识别蛋白质赖氨酸乳酸化位点的计算工具DeepKla ( http://lin-group.cn/server/DeepKla),实验结果证明 DeepKla 具有出色的预测能力和鲁棒性。并搭建了一个在线服务器,基于所提出的模型建立了一个名为DeepKla 的在线服务。DeepKla 算法的源码可在GitHub (https://github.com/linDing-group/DeepKla) 获取。
► 点击阅读
下载:1337;被引:14

Parallel-Meta Suite:跨平台可交互的微生物组快速分析套件
https://onlinelibrary.wiley.com/doi/10.1002/imt2.1
Parallel-Meta Suite(PMS)是一个易用的软件包,可以在多个平台上进行快速、全面的微生物组数据分析。PMS涵盖了广泛的数据预处理和统计方法,并提供最新的可视化结果。PMS的整个流程通过并行计算方案进行了优化,可以快速处理数千个微生物组数据。
该软件包现已在
GitHub(https://github.com/qdu-bioinfo/parallel-meta-suite)
和Gitee(https://gitee.com/qdu-bioinfo/parallel-meta-suite)
发布,其中集成了一个安装程序以实现全自动安装。
► 点击阅读
下载:2387;被引:14

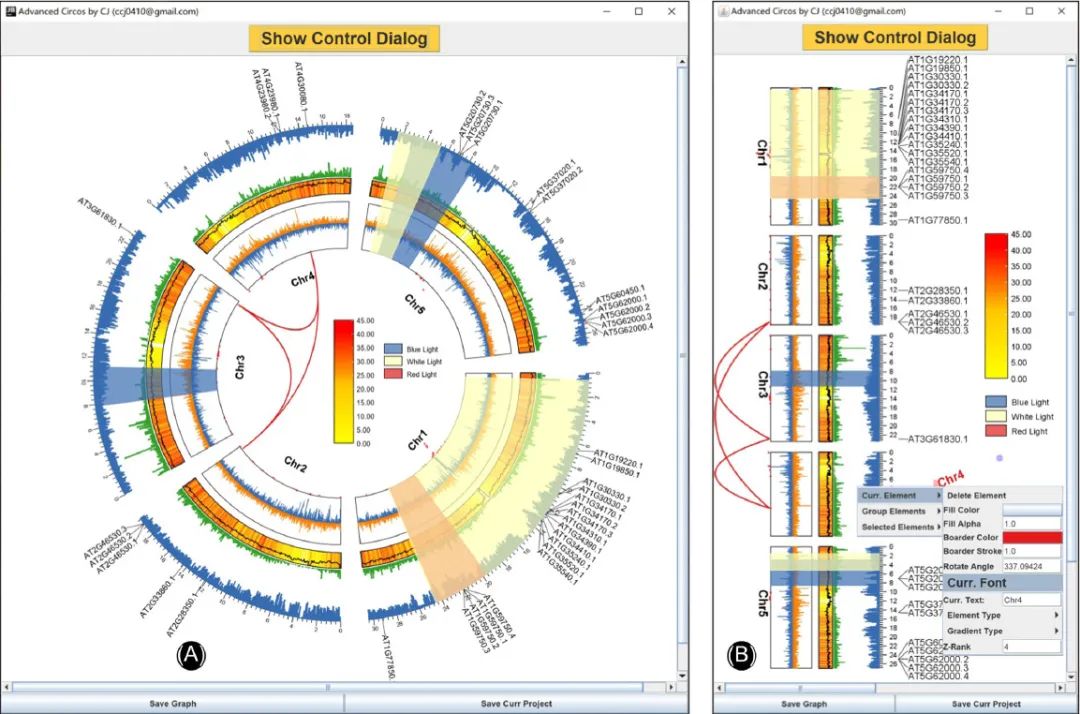
定制Circos图:使用TBtools,从数据准备到可视化
https://onlinelibrary.wiley.com/doi/10.1002/imt2.35
在该文章中,作者在TBtools中开发了“Advanced Circos”功能,提供构造Circos图的简单方法。“Advanced Circos”功能提供了一个用户友好界面,用于定制参数设置,并可用于可视化各种基因组水平数据,如基因组关联信息、比对数据、基因密度和QTL位置。
所有演示数据和TBtools的相应版本在:
https://tbtools.cowtransfer.com/s/c60a5cfec3274f
► 点击阅读
下载:5776;被引:12

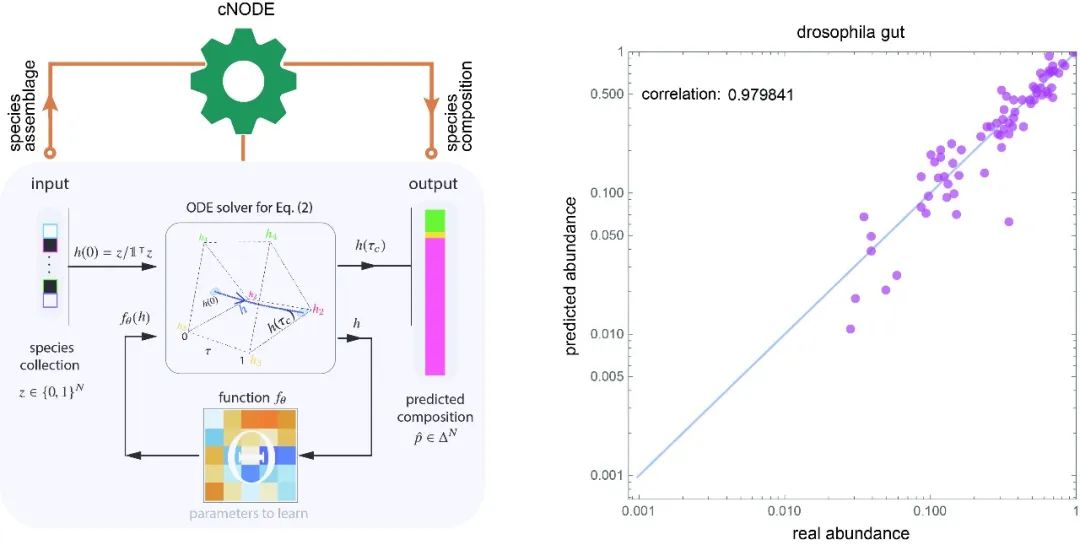
通过深度学习从物种组合中预测微生物组成
https://onlinelibrary.wiley.com/doi/10.1002/imt2.3
开发了一个深度学习框架来预测物种组合的群落组成,该框架不需要了解任何微生物动力学,各种数据的验证显示了准确的预测。
► 点击阅读
下载:3460;被引:11
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 2614
2614

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









