






微生物基因组大小和16s rRNA拷贝数-AGS和ACN
高通量测序技术的进步推动了宏基因组学研究的发展,从而可以生成大量数据。因此,宏基因组学已成为研究微生物生态学的关键。尽管如此,分析宏基因组数据是一项复杂且计算密集型的任务。通常,宏基因组由许多从许多不同物种获得的许多短读长序列组成,其中许多是未知的。平均基因组大小(AGS)和16S rRNA基因平均拷贝数(ACN)是两个重要的微生物性状,可以从未组装的宏基因组数据中计算出来,为研究微生物生态学提供有价值的信息,基因组大小与环境复杂性和生物体的生活方式有关。
宏基因组数据可以从整体上依据微生物中的35个单拷贝基因来评估数据中的基因组数量,然后根据数据中的16s rRNA基因总数除以基因组数量,即可获得平均基因组拷贝数。相关方法于2019年发表在BMC Bioinformatics上,参考文献如下:Pereira-Flores, E., Glöckner F. O., and Fernandez-Guerra A. Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data. BMC Bioinformatics. 2019;20(1):453. doi:10.1186/s12859-019-3031-y.
作者在github(https://github.com/pereiramemo/AGS-and-ACN-tools)上发布了计算AGS和ACN的两个脚本。该脚本基于未组装的宏基因组数据,能够快速对基因组大小进行计算。运行脚本依赖于docker。
AGS和ACN用法
#1. 安装,安装之前需要docker
git clone https://github.com/pereiramemo/AGS-and-ACN-tools.git #下载runacn.sh和run_AGS.sh两个脚本
#2. 拷贝到本地服务器后,可以直接使用
#2.1 acn的用法
Usage: ./run_acn.sh <input fna> <ags tsv> <input smrana> <output directory> <options>
#用法,输入文件为fastq, 默认最小16s rRNA基因长度为30。
#2.2 ags的用法
Usage: ./run_ags.sh <input fna> <input orfs> <output directory> <options>
#输入数据为ORFs fasta file来注释单拷贝基因,也可以输入fasta文件,会自动预测ORFs。
#3.1 ags具体使用参数的例子
./run_ags.sh example.fna ags_output \
--min_length 100 \
--sample_name example \
--verbose t \
--overwrite t \
--nslots 4 \
--save_complementary_data t
#3.2 acn具体使用参数的例子
./run_acn.sh example.fna example_ags.tsv acn_output \
--sample_name example \
--verbose t \
--overwrite t \
--nslots 4 \
--save_complementary_data t
4. Reference:
[1] Pereira-Flores, E., Glöckner F. O., and Fernandez-Guerra A. Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data. BMC Bioinformatics. 2019;20(1):453. doi:10.1186/s12859-019-3031-y
[2] https://github.com/pereiramemo/AGS-and-ACN-tools.gitGithup和文献信息
关于AGS和ACN的更多用法和计算原理可阅读原文, 这篇文献被ISME的一篇基于微生物生活策略的文章引用过(Chen et al., 2021 ISME J),比较新意的就是通过基因组大小和16s rRNA基因来反映微生物的生活策略。
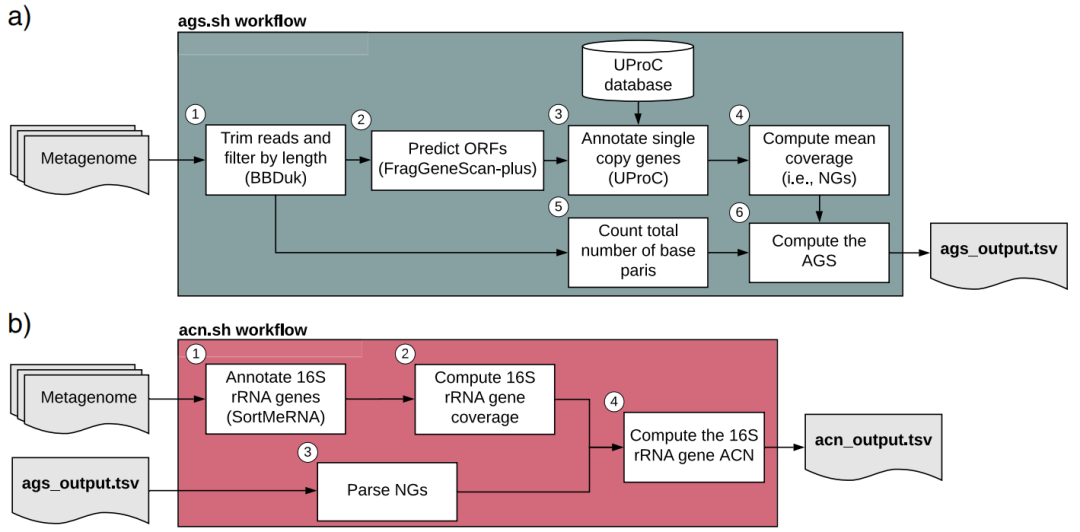
Fig. 1 Workflows implemented in the ags.sh and acn.sh tools. a ags.sh workflow consists of the following steps: 1) Filtering out and trimmingreads to obtain an appropriate read length range using the BBduk tool .
参考文献:
[1] Pereira-Flores, E., Glöckner F. O., and Fernandez-Guerra A. Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data. BMC Bioinformatics. 2019;20(1):453. doi:10.1186/s12859-019-3031-y
[2] https://github.com/pereiramemo/AGS-and-ACN-tools.git
免责声明:本文仅用于学术分享,若有侵权或错误,请联系邮箱:1372890765@qq.com删除或修改!

















 1853
1853

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









