






 中国农业科学院团队研究提出“土壤酶谱分析”方法和系列酶谱指数。研究发现酶谱指数与腐解微食物网特征密切相关,如酶化学计量比反映底物质量,胞外酶组成和多样性指示微食物网组成和多样性。该分析能反映腐解微食物网主要特征,对预测其受气候变化或人为干扰影响有重要意义。
中国农业科学院团队研究提出“土壤酶谱分析”方法和系列酶谱指数。研究发现酶谱指数与腐解微食物网特征密切相关,如酶化学计量比反映底物质量,胞外酶组成和多样性指示微食物网组成和多样性。该分析能反映腐解微食物网主要特征,对预测其受气候变化或人为干扰影响有重要意义。
点击蓝字 关注我们
土壤酶谱分析指示土壤腐解微食物网特征

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.161
● 2024年1月2日,中国农业科学院娄翼来、张晴雯团队在iMeta在线发表了题为 “Soil enzyme profile analysis for indicating decomposer micro‐food web” 的研究文章。
● 本研究提出的酶通道指数与基于微生物和线虫计算的传统分解通道指数存在密切相关性。结果表明土壤酶谱分析能够很好的反映腐解微食物网的主要特征。这对于利用土壤酶谱分析预测未来气候变化或人为干扰对土壤腐解微食物网的影响具有重要意义。
● 第一作者:邢稳, 胡宁
● 通讯作者:张晴雯(zhangqingwen@caas.cn), 娄翼来(louyilai@caas.cn)
● 合作作者:李忠芳、冯良山、张伟东、Gerhard Du Preez、张会民、李冬初、鲁顺保、Scott X. Chang
● 主要单位:中国农业科学院农业环境与可持续发展研究所、贺州学院、中国农业科学院农业资源与农业区划研究所、辽宁省农业科学院、中国科学院沈阳应用生态研究所、南非西北大学、江西农业大学、加拿大阿尔伯塔大学
亮 点

● 首次提出土壤酶谱分析可以用来指示土壤腐解微食物网特征;
● 不同酶谱的多样性指数其指示效用不同;
● 首次提出基于原生动物和酶活性计算的分解通道指数。
摘 要
在土壤腐解微食物网中,高度多样化的微生物胞外酶介导了从底物资源到多营养级微型生物类群的能量流动。本研究提出了"土壤酶谱分析"方法和一系列酶谱指数,我们假设这些指数能够反映微食物网的结构特征。基于耕地系统恢复为自然生态系统的一项案例研究,我们系统评估了与微食物网特征有关的酶谱指数的变化。我们发现,酶的碳:氮化学计量比和可分解性指数分别与底物资源的养分有效性和可分解性存在紧密的相关性。胞外酶(尤其是碳水解酶)的香浓多样性指数越高,微生物群落的多样性就越高。随着胞外酶网络复杂性和稳定性的增加,土壤微食物网的生物网络也呈现相同的增加趋势。作为代表土壤多功能性的一类参数,酶谱的总活性可以有效预测底物含量、微型生物群落的多度、多样性和网络复杂性。此外,本研究提出的酶通道指数与基于微生物和线虫计算的传统分解通道指数存在密切相关性。综合来看,这些研究结果表明土壤酶谱分析能够很好的反映腐解微食物网的主要特征。这对于利用土壤酶谱分析预测未来气候变化或人为干扰对土壤腐解微食物网的影响具有重要意义。
视频解读
Bilibili:https://www.bilibili.com/video/BV1yC4y1C7EN/
Youtube:https://youtu.be/5-ut0UWDMh4
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
土壤腐解食物网在调控陆地生物地球化学循环和维持生态系统功能方面发挥着至关重要的作用。在土壤微食物网中,细菌和真菌主要参与底物资源的分解过程。以原生动物和线虫为主要代表的微型动物通过捕食微生物从而调节了微生物种群。线虫群落物种丰富、跨越多个营养级,在土壤微食物网中处于中心位置,因此基于线虫群落数据计算的生态指数已被广泛用于描述土壤微食物网的特征。因此,由底物所支撑、由微生物和微型动物所组成的土壤微食物网研究越来越受到人们的关注(图1)。土壤微食物网的传统特征包括了底物的数量和质量、微型生物类群的多度、多样性和组成以及主要能量通道(即基于真菌和细菌的能量通道)。此外,由于生物网络的复杂性和稳定性是土壤食物网所支撑的生态系统功能的主要决定因素,也受到了研究人员的关注。
据报道,至少有500种胞外酶主要来源于土壤微生物。微生物胞外酶催化了底物降解的限速步骤并介导了能量在食物网中的流动。微生物胞外酶的催化反应具有底物专一性,而土壤底物在空间和时间上具有很大变异性,因此土壤底物在空间和时间上的变化将会影响胞外酶的分泌(图1)。酶化学计量理论认为,微生物可以通过改变所分泌的胞外酶类型来适应底物的变化,因此不同胞外酶活性的比值可以反映底物组成的变化。微生物产生的胞外酶具有身份特异性,因此土壤微生物群落组成的变化可能会导致胞外酶活性或种类的变化(图1)。此外,一些微型动物对微生物的捕食也存在偏好性,胞外酶也可能随微型动物的物种组成变化而改变(图1)。
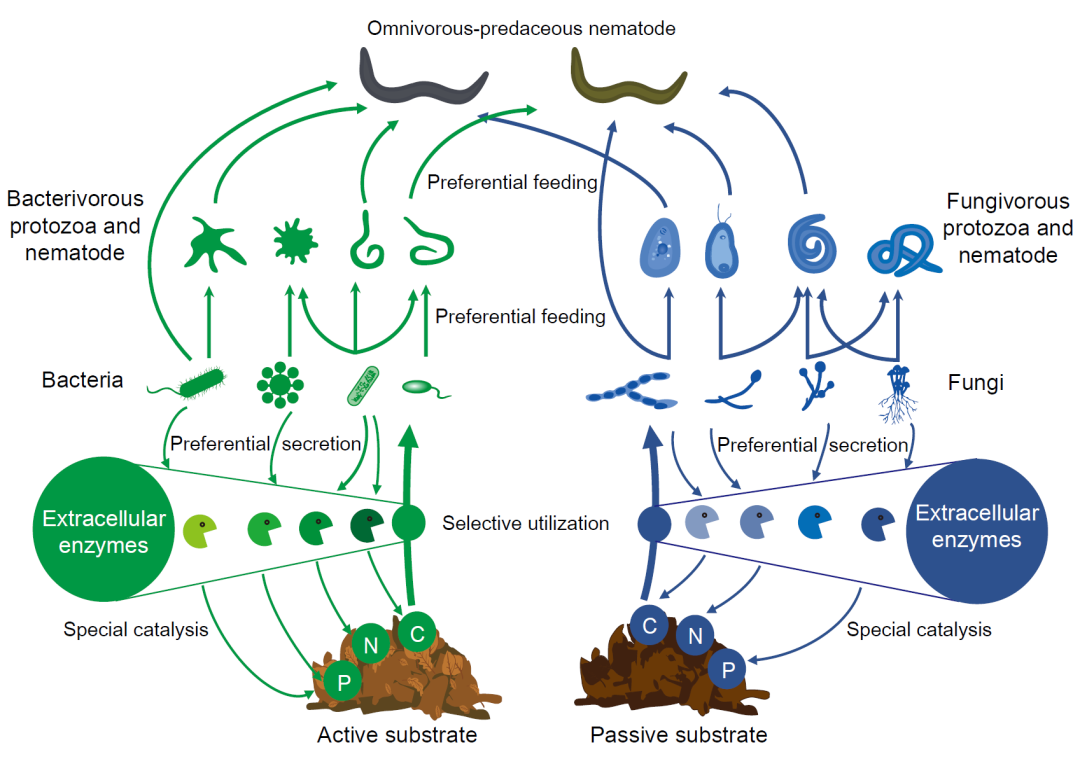
图1 土壤微食物网经典结构及其与胞外酶的机制联系
然而目前还缺乏提供腐解微食物网综合信息的生态指标。如上所述,可以通过特定的土壤胞外酶谱分析来研究微食物网的一些关键特征。本研究首次提出了一种土壤胞外酶谱分析方法,该方法有望成为微食物网特征的综合衡量指标,并能够预测土壤微食物网的状态。在本研究中,我们通过一项基于耕地系统转化为自然区域的案例研究,来评估土壤胞外酶谱分析的适用性。我们假设土壤胞外酶谱分析得出的一系列指标与农田向自然生态系统转变过程中微食物网特征的变化存在密切相关性。
结 果
腐解微食物网特征
与耕地系统相比,自然恢复处理的土壤总有机碳(TOC)、溶解性有机碳(DOC)、高锰酸钾可氧化碳(CKMnO4)、全氮(TN) 和溶解性氮(DN)含量较高(图2A)。此外,TOC:TN和DOC:DN也显著增加,而DOC:TOC和CKMnO4:TOC显著降低。在2个生长季内,各功能群和整体微食物网生物群落的组成在处理间具有显著差异性(图2B)。相比于耕地系统,除9月份的原生动物外,自然恢复处理中各功能群和整体生物类群的多度均显著提高(图2B)。各功能群和整体生物类群的丰富度和均匀度指数显著增加。除9月份的真菌香浓多样性指数外,各功能群和总体生物类群的香浓多样性指数均显著增加。与耕作系统相比,自然恢复系统的真菌:细菌(F:B)比和线虫通道指数(NCI)分别高出33.2%和48.9%,线虫通道比率(NCR)降低105%,而原生动物通道指数(PCI)在6月份降低了37.3%,但在9月份并无显著变化(图2B)。对于生物类群的共现网络(图2C),相比于耕地系统,自然恢复系统中细菌网络的网络复杂性指数例如平均度(avgK)和平均聚类系数(avgCC)值显著增加(表S1)。真菌网络的平均路径距离(GD)显著提高,而其他四类复杂性指数没有显著变化。线虫和整体微食物网网络的avgK值和GD数值均显著增加。自然恢复系统中细菌、线虫以及整体微食物网的网络鲁棒性数值显著提高,而细菌和整体微食物网的脆弱性数值则呈现降低趋势(表S1)。此外,自然恢复系统的生物网络存在更多的正相关边数、负相关边数和模块数量(表S2)。
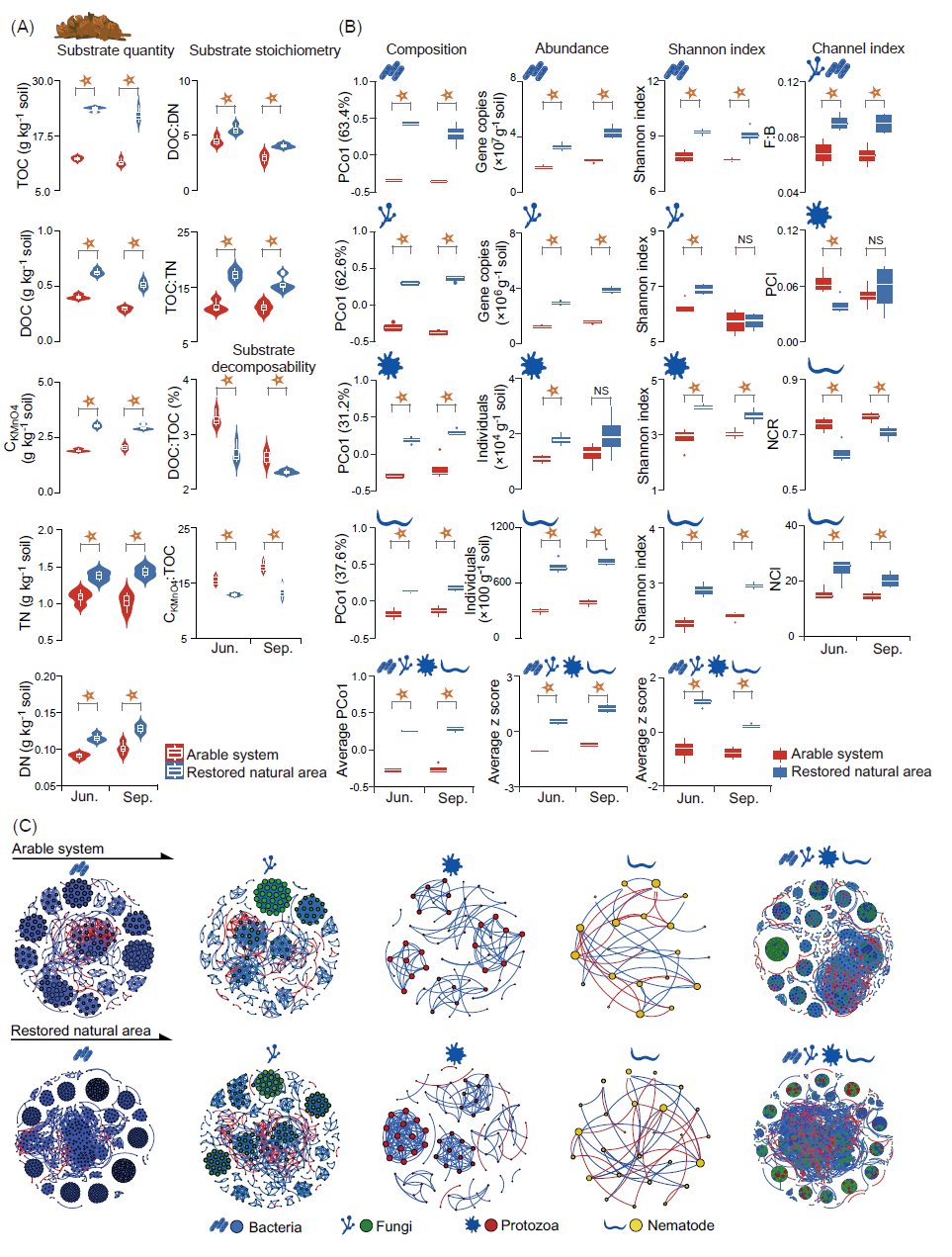
图2. 耕地系统和自然恢复系统的腐解微食物网特征
(A)土壤底物数量、化学计量比和可分解性;(B)土壤生物群落特征;(C)土壤生物网络。
酶谱指数
在两个季节,与耕作系统相比,自然恢复处理中,β-1,4-葡萄糖苷酶: β-1,4-N-乙酰-葡萄糖苷酶活性比值(BG:NAG)和β-1,4-葡萄糖苷酶: 酚氧化酶活性比值(BG:PHOX)显著降低,而β-1,4-N-乙酰-葡萄糖苷酶:亮氨酸氨基肽酶(NAG:LAP)显著增加(图3A-C)。酶谱的第一主坐标(PCo1)得分在处理间均显示出显著差异(图3D)。胞外酶总活性和香浓指数也显著提高(图3E和3F)。对于胞外酶网络,自然恢复处理的胞外酶网络复杂性数值(例如avgK、avgCC和Con)和鲁棒性数值高于耕作系统(表S3)。
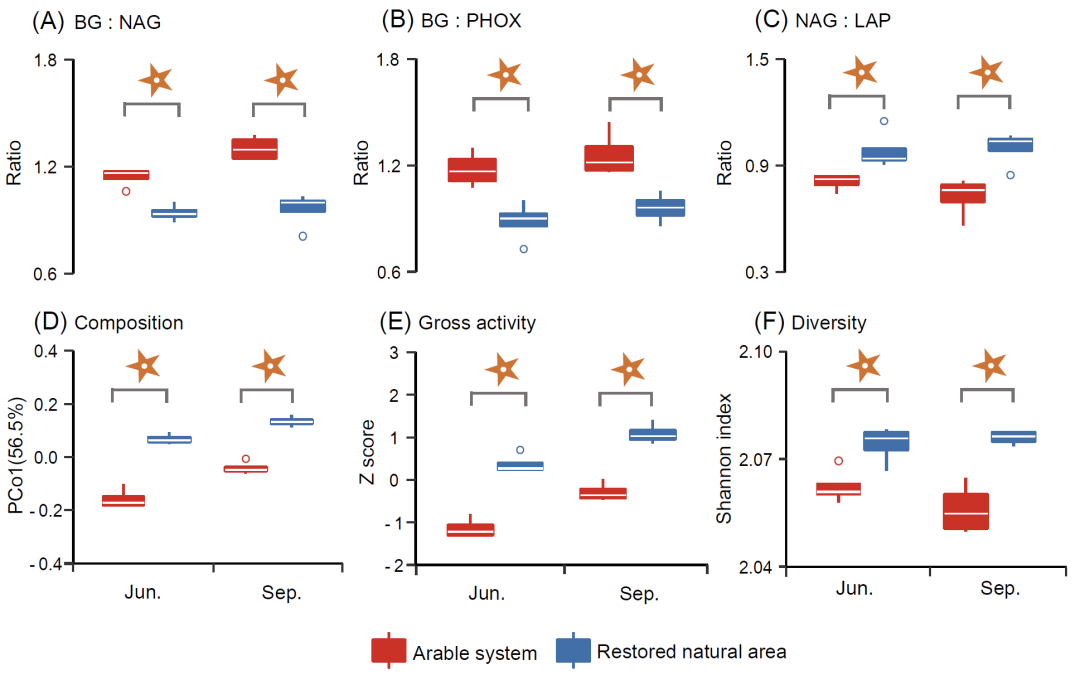
图4. 网络群落组成在盐度胁迫增加过程中的更迭
(A)BG:NAG;(B)BG:PHOX;(C)NAG:LAP;(D)组成;(E)总活性;(F)多样性。
酶谱指数与腐解微食物网特征之间的相关性
BG:NAG与底物C:N化学计量比(即TOC:TN和DOC:DN)存在显著负相关关系(图4A)。BG:PHOX与CKMnO4:TOC呈现显著正相关关系。酶谱组成与细菌、真菌、原生动物、线虫和整体食物网群落组成存在显著相关性(图4A)。酶谱的香浓指数(包括选定的所有8种酶)与各功能群(真菌除外)和整体生物群落的香浓指数存在显著正相关关系。碳降解酶、碳氮水解酶和碳水解酶谱的香浓指数与细菌、原生动物和线虫以及整体生物群落的香浓指数也存在显著正相关关系。其中,碳水解酶与生物群落的相关性最强。酶网络的avgK值与细菌网络的avgK数值存在正相关关系(图4B)。酶网络的avgCC值与细菌网络和整体生物网络的avgCC值存在显著的正相关关系。此外,酶网络的鲁棒性与细菌网络的鲁棒性数值呈现正相关关系。胞外酶总活性与底物数量(即TOC、DOC、CKMnO4、TN和DN含量)、各功能群和整体生物群落的多度、各功能群(真菌除外)和整体生物群落的香浓指数呈正相关关系(图4A)。胞外酶的总活性与细菌、真菌、原生动物和线虫网络的正相关边数、负相关边数和模块数量也呈正相关关系(图4C)。此外,NAG:LAP与F:B和NCI呈正相关,与NCR呈负相关,而与PCI没有显著相关性(图4A)。
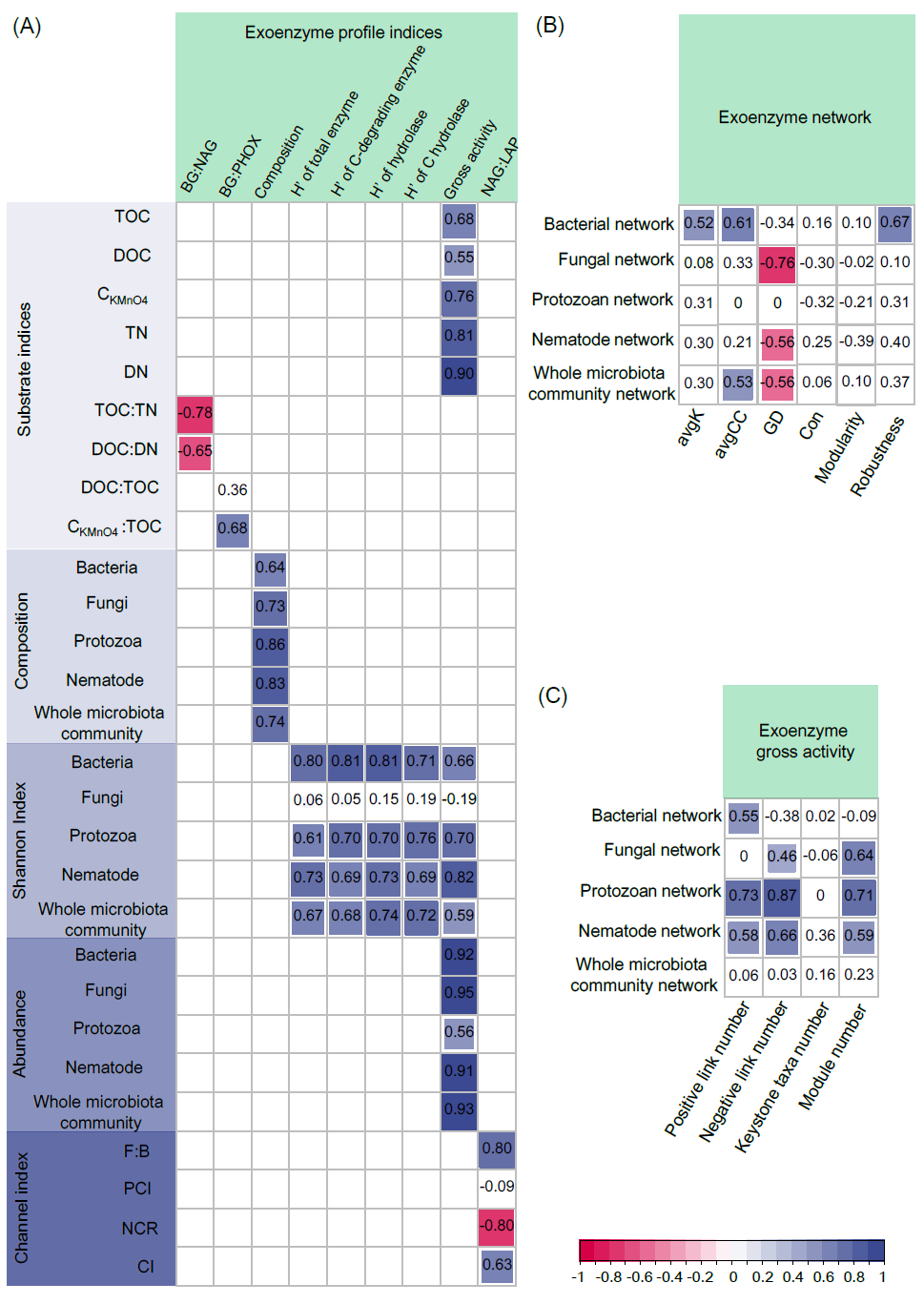
图4. 土壤酶谱指数与腐解微食物网特征之间的相关性
(A)土壤酶谱指数与土壤底物和生物群落特征之间的相关性;(B)酶谱网络和生物网络指数的相关性;(C)酶总活性和生物网络指数的相关性。
讨 论
胞外酶的化学计量比和碳可分解性指数指示底物质量变化
底物的化学计量比和底物可分解性是土壤微食物网功能的关键驱动因素。在我们的案例研究中,在耕地系统转化为自然系统后,TOC:TN和DOC:DN比值都增加了,这表明土壤微食物网的碳有效性增加,但底物的氮素有效性降低。CKMnO4:TOC和DOC:TOC比值降低,表明底物碳的可分解性降低。我们发现在自然恢复处理中,BG:NAG比值降低,并与TOC:TN和DOC:DN呈负相关。这表明了BG:NAG在评估底物C:N化学计量方面的有效性。这些研究结果有力地支持了酶化学计量理论,即土壤微生物可以通过改变分泌的胞外酶类型来适应营养物质缺乏的环境,从而促进营养物质循环,从而加速养分周转。虽然本研究仅研究了底物C:N化学计量比,但许多研究也表明胞外酶的C:P或N:P分别与底物C:P和N:P之间存在密切联系。此外我们观察到在耕地系统转换为自然生态系统后,BG:PHOX显著降低,并与CKMnO4:TOC和DOC:TOC呈显著正相关关系。这表明基于酶活性计算的碳可分解性指数可以指示底物碳的可分解性,并且这与先前的研究报道了微生物可以调节资源向胞外酶的相对分配结果一致。
本研究选择了8种胞外酶来指示土壤微食物网的相应指数,是由于在大多数研究案例中它们被认为催化了相关底物复合物中含量最高的底物组分。然而,这些指数的效用可能取决于特定的底物。例如先前的一项研究发现,在以纤维素为主要碳源的研究中,BG:NAG可以有效地反映微生物对碳氮的相对需求;而在以几丁质、肽聚糖和蛋白质为主要碳源的研究中,BG:NAG不能用于指示微生物对碳和氮的相对需求。因此,根据存在的特定底物适当选择特定的胞外酶,对于提高这些酶指数指示底物化学特征变化的效用来说至关重要。
胞外酶的组成和多样性指示微食物网组成和多样性
与先前多项研究的结果一致,我们发现,与耕地系统相比,自然恢复处理的每个功能群和整体生物群落在丰富度、均匀度和香浓指数方面表现出更高的多样性。这可能是由于自然恢复系统中底物数量的增加(例如本研究中所述的TOC和TN的增加)、底物复杂性的增加,栖息地的改善(例如土壤pH和团聚体的改善)更有利于维持多种微生物的共存。然而,对于在两种处理下检测到的所有类型的胞外酶,耕地和自然恢复系统之间的胞外酶丰富度没有差异可能是由于我们的研究只包含了八种酶并且这些酶都是常见的。选择更多数量的其他胞外酶,并且纳入稀有的胞外酶,对于提高酶谱的指示效用可能是重要的。正如我们所预期的,在自然恢复生态系统中,胞外酶组成发生了显著变化,香浓指数也更高。胞外酶的组成和香浓指数,与各生物类群和整体生物群落的组成和香浓指数之间也存在显著相关性。这表明了胞外酶谱的多样性指示效用,首先可以归因于不同种类的微生物执行不同的代谢功能,并分泌不同类型的胞外酶。此外,微生物-食微动物(原生动物和线虫)的营养级相互作用也解释了胞外酶和食微动物之间的多样性关系。在本案例研究中,微生物和食微动物多样性之间存在显著的正相关性(图S4)。一方面,微生物群落结构可以通过上行效应改变食微动物群落结构。因此,胞外酶可以与食微动物群落结构产生间接联系。另一方面,食微动物群落结构可以通过下行效应调节微生物群落结构,进而间接调控微生物分泌的胞外酶谱结构。
如前所述,在我们的案例研究中,底物化学计量学和可分解性的变化,导致特定胞外酶结构在自然恢复区域下发生显著变化。这可能导致胞外酶谱均匀度在指示生物类群多样性方面的实用性上令人感到困惑。为了最大限度地减少这种影响,我们进一步选择了胞外酶谱的特定子集来评估生物类群的多样性指标。我们发现,碳水解酶的香浓多样性指数与大多数生物群落和整个微食物网的香浓多样性指数之间存在强烈正相关关系。这可能是因为与碳水解酶谱相比,所研究的总酶、碳降解酶、水解酶谱是由更多类型的胞外酶组成,因此在所研究的酶谱中,碳水解酶谱受底物化学计量和可分解性的影响最小。这些研究结果表明,当使用胞外酶均匀度来评估生物类群多样性时,选择合适的胞外酶类型对于研究领域和研究问题很重要。或者,选择更多数量的胞外酶可能有助于提高这种方法的有效性。
胞外酶谱的网络复杂性和稳定性指示微食物网的网络复杂性和稳定性
传统上,土壤食物网的复杂性是基于生物类群之间的营养级相互作用来描述的。以物种为节点,以物种-物种关系为纽带的网络通常用于表征土壤食物网中复杂的生态相互作用,例如捕食、竞争和互惠。利用这种网络分析,相关研究开发了一系列量化生态网络复杂性和稳定性的参数。由于不同微生物类群之间的胞外酶分泌模式可能不同,因此微生物类群之间的相互作用会诱导相应胞外酶的某些相关性。例如,假设胞外酶A1(随机命名)来源于微生物类群B1,胞外酶A2来源于分类群B2,那么B1和B2之间的相互作用(假设为正向关系)将导致A1和A2之间的统计学正相关性。更高的土壤食物网生物多样性往往支持更复杂的生态网络,而更复杂的网络通常表现出更高的稳定性。这可能是因为网络的连通性和模块性等复杂性指标与鲁棒性等稳定性指标呈正相关关系。在本案例研究中,我们还发现,与耕地系统相比,随着自然恢复处理中生物类群多样性的增加,土壤微食物网的网络复杂性和稳定性也增加,正如其更高的avgK、avgCC和GD数值,网络鲁棒性的增加,网络脆弱性的降低所示。此外,尽管共现网络分析中仅包括八种类型的胞外酶,但我们观测到了自然恢复处理网络中较高的avgK、avgCC、Con以及更高的鲁棒性数值。这表明,与耕地系统相比,自然恢复系统中存在更复杂和更稳定的胞外酶网络。并且胞外酶网络的avgK和avgCC分别与生物类群网络的avgK和avgCC也呈现出正相关关系。
胞外酶总活性指示底物数量、生物群落多度、多样性和网络复杂性
众所周知,土壤微生物通过分泌胞外酶催化底物来获得生长所需的能量、碳和多种营养物质(图1)。通常,酶活性是反映土壤肥力和微生物功能的关键指标。在我们的案例研究中,土壤有机碳和氮含量的增加,刺激了微生物的生长,进一步提高了胞外酶的总活性。此外,微生物通过上行效应进一步促进了捕食者(即原生动物和线虫)的生长,导致原生动物和线虫的多度显著增加。因此,胞外酶的总活性不仅与土壤碳氮含量呈正相关,还与微生物、原生动物和线虫的多度呈正相关。这表明胞外酶谱的总活性可以作为反映微食物网底物数量和生物群落多度的指标。
由于胞外酶谱的总活性反映了土壤微生物的多种代谢功能(即土壤多功能性),并且土壤生物群落维持多功能性的能力强烈依赖于土壤生物多样性,我们假设胞外酶总活性与土壤生物多样性之间会存在很强的关联性。事实上,我们观察到多种土壤生物(即细菌、原生动物、线虫和整体生物群落)的香浓多样性指数与胞外酶谱总活性呈正相关。这可以用经典假设“互补效应”和“选择效应”来解释。互补效应被定义为不同物种各自利用不同生态位的能力,其结果是多物种组合将比任何单一物种利用更多的资源。因此,不同物种的组合不仅可以促进物种的共存,而且比单独生存的任何物种都要好。选择效应表明具有特定性状特征的优势物种可能在群落中过度发挥特定的功能。除了物种丰富度外,均匀性也被认为有利于促进群落的多功能性。在一个由少数物种主导的不均衡群落中,任何生物都可能与其近属物种产生相互作用。相反,在物种均匀度高的群落中,一种生物很可能与其他功能不同的生物相互作用,这有助于促进协同作用从而提高土壤的多功能性。
生物类群之间的关联复杂性为理解生物多样性对多功能性的积极影响提供了一个新的维度。因此,正如我们的案例研究所表明的那样,胞外酶谱的总活性也可以反映微食物网的网络复杂性。这可以通过以下三个方面来解释。首先,不同物种类群之间的正向(如促进)和负向(如竞争)相互作用驱动生态系统功能。如本案例研究所示,更复杂的网络可能具有更多数量的正相关关系和/或负相关关系(表S1),这表明可能存在更多的物种相互作用,这可能会对土壤酶总活性产生积极影响。此外,关键类群是具有高度连接性的分类群,能够赋予群落更大的生物连通性。在我们的研究中,在自然恢复处理的网络中发现了更多的关键物种分类群。这些关键物种类群对维持网络结构和多种功能很重要,这可能对土壤的多功能性产生积极影响。网络中的模块是一组高度连接的类群。在食物网中,功能不同的群体往往形成不同的网络模块,因此包含更多模块化结构的群落可能具有更多的功能群多样性和更强的提供多种生态系统功能的能力。事实上,在我们的案例研究中,在将耕地系统转换为自然恢复区域后,检测到了更多数量的网络模块。
胞外酶通道指数反映微食物网分解通道结构
土壤食物网中的能量流动主要由基于细菌和基于真菌的通道所驱动。这两个通道的相对重要性是预测食物网功能和可持续性的一个关键参数。正如我们所预期的,相对于耕地系统,自然恢复生态系统的分解通道转向以真菌为优势的通道。F:B和NCI的增强以及NCR的减少证明了这一点,这与前人的报道一致。传统上,原生动物被认为主要捕食细菌。最近的研究指出,一些原生动物(如A. castellanii)更喜欢取食真菌。在此基础上,我们首次尝试提出PCI来指示不同生态系统之间的分解通道结构变化。然而在本研究中,与耕地系统相比,在自然恢复生态系统的PCI值没有增加。这表明上述关于某些原生动物偏好取食真菌的观点可能值得怀疑,因此需要在未来的研究中对PCI的计算做进一步改进。或者,这可以通过本研究中有限的采样时间点来解释。就PCI作为一个瞬时指标而言,其对自然恢复处理的响应可能具有季节性特点,并且该响应可能无法有效地指示某些特定取样时间点上总体分解通道结构的变化。因此,仍需对PCI实用性做进一步的检验,包括采纳更多的取样时间点。
考虑到NAG主要来源于真菌,我们首先提出了由NAG与LAP的比值计算的酶通道指数。这两种酶参与了氮循环。在本研究中,NAG:LAP的比值,在耕地转换为自然生态系统后显著增加,并与F:B和NCI呈强烈正相关性,与NCR呈负相关性。这表明了NAG:LAP比值在反映分解通道结构方面的有用性。NAG:LAP越高,意味着基于真菌的能流通道在土壤微食物网中的相对重要性越大。微生物对土壤食物网的能量流动起主要作用,因此F:B通常被作为指示分解通道结构的基本指数。由于高营养级生物(如线虫)的繁殖期较长,在基于线虫丰度计算的通道指数的响应可能会有几天到几周的滞后时间。相比之下,微生物产生的胞外酶可以随着环境变化而迅速响应。因此,基于酶活性计算的通道指数在预测分解通道结构的即时变化方面可能比基于动物丰度计算的通道指数更为有效。
结 论
在这项关于耕地系统转变为自然恢复生态系统的案例研究中,我们首次提出的土壤酶谱指数与腐解微食物网的相应参数之间存在密切关联性。我们的研究结果表明“土壤酶谱分析”可以作为综合反映腐解微食物网特征的有用工具。然而土壤酶谱分析的效应还需要进一步研究。此外,我们建议相关研究考虑收集足够数量和适当类型的胞外酶。
为了优化和开发酶谱指数并促进“土壤酶谱分析”在指示土壤微食物网变化方面的应用,未来的工作需要进一步加强以下两个研究方向。(i)土壤胞外酶的具体来源:尽管已经发现了许多土壤胞外酶,并且已知它们主要来源于微生物,但我们对胞外酶的特异性来源了解仍然有限。这将有助于我们更好地理解胞外酶和微食物网之间的理论联系。例如,如果某些胞外酶特异性来源于土壤微型动物,那么此类胞外酶的相对活性有望指示微食物网的垂直营养级结构;(ii)胞外酶活性的高效测量:荧光检测技术大大提高了土壤胞外酶活性的测定效率,但该技术可以测定的酶类型仍然有限。如上所述,“土壤酶谱”中纳入的酶特性越多,其对微食物网的多样性和网络参数的预测能力就越强。
方 法
研究地点
本研究选择了一项长期田间试验的“耕作系统”和“自然恢复系统”处理。该试验于1990年在湖南省祁阳国家土壤质量观测实验站建立。该地区属于亚热带季风气候,年平均降水量为1250mm和年均温为18°C。田间试验包括一系列关于土地利用和养分管理策略的处理,每个小区面积为200 m2。在田间试验开始之前,表层土壤(0-20cm)的化学性质如下:TOC=6.7 g kg–1,TN=1.07 g kg–1,有效氮(AN)=79 mg kg-1,有效磷(AP)=10.8 mg kg-1,pH=5.70。
试验设计
本研究选择了其中两种处理,即传统耕作系统(即耕地)和自然恢复区域。每个处理设置4个小区作为重复,每个小区面积为25m2。耕地系统采用雨水灌溉的小麦-玉米轮作模式,施肥方式为常规施肥。在玉米季,尿素、过磷酸钙和氯化钾施用量分别为188 kg N ha-1 yr-1、41 kg P ha-1 yr-1和78 kg K ha-1 yr-1。在小麦季,尿素、过磷酸钙和氯化钾施用量分别为165 kg N ha-1 yr-1、36 kg P ha-1 yr-1和68 kg K ha-11 yr-1。自然恢复区域内的植被以草本植物、灌木和乔木为主。
土壤取样
于2021年6月和9月采集土壤样本。每次取样时,从每个小区中采集六个表层土壤(0-15cm)样本,混合后形成一个复合样本。该复合样本被分成三份,并使用相应网目尺寸的网筛过筛。一部分土样保存在-80℃,被用于提取土壤基因组DNA,另一部分土样风干后被用于测量土壤性质。最后,剩下的一部分土样在4℃下保存,一周内用于测定土壤水分和胞外酶活性以及提取原生动物和线虫。
土壤底物分析
采用元素分析仪(Vario EL V, Elementar, Germany)测定总有机碳(TOC)和总氮(TN)含量。由于原位土壤呈酸性,其碳酸盐含量可以忽略,因此我们将全碳含量作为土壤总有机碳(TOC)的近似值。高锰酸钾氧化的碳用来指示易降解有机碳(CKMnO4),使用0.5 M K2SO4溶液提取土壤溶解性有机碳(DOC)和溶解性氮(DN),上清液经0.45 µm过滤器过滤,采用自动TOC/TN分析仪(multi‐N/C 3000,Analytik Jena)测定滤液中的C和N浓度。TOC:TN和DOC:DN被用来计算底物C:N化学计量比。DOC占TOC的比例以及高锰酸钾氧化的碳占TOC的比例(%)用来评估土壤有机碳的可分解性。
土壤细菌和真菌分析
按照制造商的说明,使用FastDNA SPIN土壤试剂盒(MP Biomedicals,Santa Ana,CA)从0.5g土壤样本中提取土壤基因组DNA。使用StepOnePlus实时荧光定量PCR系统,通过定量PCR方法测定了细菌和真菌核糖体RNA(rRNA)基因拷贝数。引物27F/519R和ITS1/ITS4分别用于靶向16S和ITS rRNA基因。利用Illumina MiSeq测序平台,对所有纯化后的扩增子进行双末端PE300测序。细菌16S rRNA基因引物为338F/806R,真菌ITS1 rRNA基因引物为ITS1F/ITS2R(Quantitative Insights into Microbial Ecology 2,2021.4版本)处理测序数据。使用dada2插件对序列进行质量过滤,去噪和去除嵌合体,然后聚类生成扩增子序列变体(amplicon sequence variants,ASVs)。依据所有样本中的最小序列数量,对所有样本进行抽平,并计算了包括香浓指数在内的α多样性指标。参照SILVA(132版本)和UNITE(8.3版本)数据库,使用feature-classifier插件中的classify-sklearn Naive Bayes分类器,分别对细菌和真菌序列进行分类学鉴定。
原生动物分析
使用最大似然法测定了土壤原生动物主要类群鞭毛虫、变形虫和纤毛虫的密度。将2 g新鲜土壤悬浮在18 mL无菌水中,180 rpm充分振荡30分钟。一式四份,将三倍稀释系列的悬浮液加入到96孔微量滴定板中。将滴定板放置于黑暗25°C条件下进行培养。4天、7天和11天后,使用倒置光学显微镜(Nikon Eclipse TS100)观察微孔中是否存在原生动物。原生动物的密度单位为每克干土中的个体数量。为了进一步描述原生动物群落,基于引物TAReuk454FWD1/TAReukREV3扩增了18S rRNA基因并进行了高通量测序。原生生物群落的测序数据处理过程与细菌、真菌的测序数据处理过程相似。参考PR2数据库(4.14版本)对序列进行分类鉴定。由于本研究只关注原生动物,因此参照相关文献,采用半定量分析方法,从获得的ASV表格中筛选了属于原生动物的序列,然后计算了包括香浓指数在内的alpha多样性。参照之前的研究,我们将ASVs归类为食细菌和食真菌类群。本研究首次提出以食真菌原生动物与食细菌原生动物相对多度之比计算的PCI作为一个新的分解通道指数。
土壤线虫分析
采用浅盘法,从每个样品的100g新鲜土壤中提取线虫,在65°C下杀死并用4%甲醛固定。使用显微镜(Olympus BX50)对提取的线虫进行计数并鉴定到属水平。线虫群落被划分为4个营养类群:食细菌线虫、食真菌线虫、捕食-杂食线虫和植食性线虫。利用腐解微食物网中包含的前3个营养类群(即自由生活线虫)进行线虫群落分析,从中选取至少100个线虫计算多样性指数(即丰富度、Pielou均匀度指数和香浓指数)。使用抽平后的数据进一步计算NCR:NCR=B/(B+F),其中B和F分别是食细菌线虫和食真菌线虫的多度。此外,根据生活史特征将线虫划分为不同的功能群。有关线虫功能群分类的更多信息,可访问http://nemaplexucdavisedu/。线虫通道指数NCI是分解通道(细菌vs.真菌)的衡量标准,计算方法如下:NCI= 100×(0.8×Fu2/(3.2×Ba1 + 0.8×Fu2)),其中Ba1和Fu2分别是食细菌线虫c-p1和食真菌线虫c-p2类群中线虫的多度。
土壤微型生物总体特征
基于各类群的相对多度,使用R(4.1.1版)中的vegdist函数分别计算了细菌、真菌、原生动物和线虫群落的Bray-Curtis相异性矩阵。然后对各处理进行主坐标分析,以第一主坐标得分(PCoA1)作为各生物群落组成的指标。将四类生物的PCoA1得分的平均值作为整体土壤生物群落组成的指标。然后采用z-score标准化方法,将各类群其标准化后的多度、丰富度、Pielou均匀度指数和香浓指数的平均值分别作为整体土壤生物群落多度和多样性的指标。
微型生物共现网络分析
基于生物类群的相对多度,网络分析方法用来探索土壤细菌、真菌、原生动物、线虫、整体土壤生物群落的所有个体成员之间的潜在相互作用。基于细菌、真菌、原生动物的ASVs和线虫属,构建了土壤生物群落的相对多度表格,进一步分别构建了耕地系统和自然恢复系统的土壤微食物网网络。首先计算Spearman相关矩阵,相关系数绝对值的截断值为> 0.6, P值< 0.05。其次,使用Gephi软件对相关网络进行可视化。为了评估网络的复杂性,我们使用igraph包计算了5个网络拓扑学参数:平均度(avgK)、平均聚类系数(avgCC)、平均路径距离(GD)、连通性(Con)和模块化(modularity)。模块化反映了食物网中各成员之间的高度协同程度。此外,鲁棒性(robustness)和脆弱性(vulnerability)用来评估网络稳定性。鲁棒性定义为在随机或将目标节点移除后,网络中剩余成员的比例。脆弱性定义为每个节点对全局效率的相对贡献,其中全局效率是指信息在网络中传播速度的快慢。本研究计算了当去除50%随机节点时每个网络的鲁棒性,并参考Yuan等(2021)计算了网络中节点的最大脆弱性。还计算了正负相关边的数量。最后,根据Zi和Pi的阈值确定网络中单个节点的拓扑作用。节点被分为四类:module hubs (Zi>2.5)、network hubs (Zi>2.5和Pi<0.62)、connectors和peripherals(Zi<2.5和Pi<0.62)。connectors、module hubs和network hubs被归类为关键节点。
胞外酶活性分析
分析了四种碳水解酶(α-1,4-葡萄糖苷酶,AG;β-1-4-葡萄糖苷酶,BG;纤维素酶,CB;木糖苷酶,XS和两种氮水解酶(β-1,4-N-乙酰氨基葡萄糖酶,NAG;亮氨酸氨基肽酶,LAP)和两种氧化酶(酚氧化酶,PHOX;过氧化物酶,PEOX)的潜在活性。将2.75g(用于水解酶)或1g(用于氧化酶)的新鲜土壤加入到91 mL的50mM乙酸钠缓冲液中,然后使用磁力搅拌器充分搅拌1min。乙酸钠缓冲液的pH值调整到土壤样品的近似pH值。然后将土壤悬浮液(200μL)加入96孔黑色微板中,每孔均包含200mM荧光底物(50 μL)。每个土壤悬浮液同时添加一系列浓度的4-羟甲基-7-香豆素(MUB)或7-氨基-4-甲基香豆素(MUC),获得标准曲线。在微孔板中,每个样品均包含6次技术重复。将微孔板放置于25°C的黑暗环境中培养3小时。使用荧光光谱仪(Beckman Coulter DTX 880),在365nm激发和450nm发射波长下测定荧光值。对于PHOX,将150 μL的土壤悬浮液加入到96孔透明微板中,每孔添加25 mM 50 μL左旋多巴(L-DOPA)和5 mM 50 μL的EDTA。阴性对照中只添加150 μL土壤悬浮液和100 μL醋酸钠缓冲液。对于PEOX,额外加入15 μL的0.3% H2O2。将微孔板放置于在25°C黑暗环境中培养20小时,然后在450 nm下测定吸光度。所有酶活性的单位为nmol g−1 soil h−1。
酶谱分析
计算了BG:(NAG+LAP)和BG:PHOX分别表征底物的化学计量比和可分解性。NAG:LAP用来分析土壤微食物网中真菌分解通道相比于细菌分解通道的优势变化,这是由于NAG被认为主要是由真菌分泌,而细菌是LAP的主要分泌来源。计算了包含了8种酶(4种碳水解酶、2种氮水解酶和2种氧化酶)的群落组成。与土壤生物群落组成的计算方式一致,根据8种酶各自的比例,通过计算酶群落的Bray-Curtis相异性矩阵进行主坐标分析。将PCo1得分作为酶群落组成指标。由于观测到的8种酶活性的数量级之间存在较大差异,我们在计算群落组成之前将每种酶的原始数据除以其在所有样本中观测到的最大值,然后乘以100。将标准化后的所有酶活性的平均值作为整体酶群落的总活性指数。由于8种酶活性在每个样本中均能够被检测到,本研究没有计算丰富度,而是使用公式H=(-∑Pi[lnPi])计算了香浓多样性指数,其中Pi是指某一具体酶活性占所有酶活性之和的比例。然后计算了整个酶谱(本研究测定的所有8种酶)、碳降解酶(碳水解酶和氧化酶)、水解酶、碳水解酶的香浓指数。最后,参照生物群落网络构建方法,基于酶的相对活性,分别构建了耕地系统和自然恢复系统的酶谱网络,并计算了其复杂性和稳定性指数。
统计分析
采用T检验,分别比较了相比于耕作系统,自然恢复处理对土壤生物群落特征和酶谱指标的影响。Pearson相关分析用来评估土壤酶谱指数与生物群落特征之间、土壤微生物(细菌和真菌)与底物以及更高营养级生物之间的关系。
引文格式:
Xing, Wen, Ning Hu, Zhongfang Li, Liangshan Feng, Weidong Zhang, Gerhard Du Preez, Huimin Zhang, Dongchu Li, Shunbao Lu, Scott X. Chang, Qingwen Zhang, Yilai Lou. 2023. “Soil Enzyme Profile Analysis for Indicating Decomposer Micro‐Food Web.” iMeta e161. https://doi.org/10.1002/imt2.161
作者简介

邢稳(第一作者)
● 现为中国农业科学院农业环境与可持续发展研究所博士后,博士毕业于中国科学院植物研究所。
● 研究方向为土壤微食物网及碳氮循环。相关学术成果发表于iMeta, Soil Biology and Biochemistry和Functional Ecology等期刊。参编著作1部,担任Microorganisms期刊客座编委。

张晴雯(通讯作者)
● 中国农业科学院农业环境与可持续发展研究所研究员,农业农村部农业农村生态环境重点实验室副主任,中国农业科学院农业清洁流域创新团队首席,生态安全研究室主任。
● 主要开展流域源汇特征及环境效应、碳氮循环及驱动机研究。主持国家自然科学基金3项,主持国家科技重大专项课题、国家重点研发课题等16项。参加完成的项目有国家自然科学基金西部重大研究计划项目、中国科学院“引进国外杰出人才”、国际原子能机构项目、中德政府国际合作项目等20多项。出版专著5部,发表科技论文88篇,授权发明专利12项,编制地方标准5件,软件著作权10件。获陕西省科学技术一等奖2项、省部级二等奖3项。研究成果入选中国科协2019年度“中国生态环境十大科技进展”。

娄翼来(通讯作者)
● 中国农业科学院农业环境与可持续发展研究所研究员。
● 主要从事土壤微食物网与碳氮循环、气候智慧农业等方面研究,主持国家自然科学基金、国家重点研发计划等项目课题,以第一/通讯作者在Soil Biology and Biochemistry、Biology & Fertility of Soils、Catena等期刊发表论文20余篇,主编《喀斯特生态恢复土壤微食物网》等著作,荣获“我国农田土壤有机质演变规律与提升技术”等省部级科技进步奖,担任Climate Smart Agriculture执行主编。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法


1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、Scopus等数据库收录。
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 580
580

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









