






目录
(一)BASALT支持的数据类型及特点
(二)依赖程序:
1)系统及脚本语言
2)内置的自动分箱软件
3)映射、解析、预测及比对软件
4)组装及碱基修正软件
5)基因组评估软件
6)工作站硬件要求
(三)安装方法
1)在外网或翻墙的情况下,使用BASALT_setup_international.py安装
2)在内网且无法翻墙的情况下使用BASALT_setup_China_mainland.py安装
3)手动配置BASALT环境(国内用户推荐用该方式安装)
4)利用singularity运行预安装好的BASALT
5)测试文件
(四)软件使用
1)参数简述
2)使用范例
3)使用举例
(五)安装和运行中常见问题
(六)常见其他问题(FAQ)
(七)软件引用
(一) BASALT支持的数据类型及特点
BASALT是一个多功能工具包,支持从(1)纯二代序列的组装序列、(2)纯三代序列组装序列(现阶段主要支持PacBio-HiFi数据,其他纯三代数据如ONT因覆盖率计算的问题仍在评估中,约在稍晚开放支持)及(3)二代加上三代数据的混合组装的一系列数据中获取高质量MAGs。BASALT具有以下几个的特点:
1)BASALT支持多组装文件的相互精修
已有的软件如MetaWRAP及DASTool仅支持单一组装文件的分箱,并在多组装文件出现时利用去冗余软件,如dRep进行去冗余。部分分箱软件,如VAMB等,也具有多组装文件共同输入的功能,但我们的实测结果表明这些软件做多组装文件时的分箱效果,不如它们做单一分箱的效果。在总结我们过往的测试结果的基础上,BASALT开发了一套通过多个组装文件同时分箱后,利用我们自主开发的覆盖率一致性算法识别冗余基因组,挑选质量最好的基因组,并且进一步将冗余基因组对选定的最好质量的基因组进行修正。这里面包括通过冗余基因组中存在的片段补全被选定的基因组,及利用冗余基因组对选定基因组进行补洞(gap)。故理论上使用了BASALT后不需要再用dRep进行去冗余,但用户可根据自己的需求进行修改。
2)基于深度学习的修正(refinement)模块可大幅度的提高基因组质量
结合自主开发的神经网络的算法,BASALT可高效率的识别潜在错误片段,并进过自动评估后去除潜在的错误片段。在过去我们对Binning refinner和RefineM评估中,BASALT的修正效果均大幅度好于这两个软件。由于metaWRAP内置的修正软件所用的是Binning refiner, metaWRAP在修正上差于BASALT。DASTool与BASALT比较的结果也显示BASALT修正的效果好于DASTool。并且,这一模块可独立使用,如用户有一组已经完成分箱的基因组,可通过BASALT内的data feeding脚本将基因组和原始数据提供于BASALT,进行覆盖率等计算,随后用refinement模块进行修正。
3)BASALT对polish流程及重组装进行大幅度优化
在二代及三代数据存在的情况下,BASALT强化了修正功能,及我们整合了利用pilon等软件的polish的功能。在这一过程中,BASALT进行了大量polishing的效率优化,在运行相同的polish次数(run)的情况下,BASALT可以比用户单独使用polish软件节省约90%的polish的时间。BASALT对各种情况下的数据进行重组装的优化。尤其是对2/3代混装的效率和质量均达到了已知最好的水平。纯3代数据的优化仍在进行中,该功能会在稍晚释放给用户使用。我们仍需提醒用户一点,即重组装可大幅度提高基因组质量,但同时也会导致获取基因组的时间增长非常多。我们在2+3代混合重组装时测试了Opera-MS/Unicycler及SPAdes等软件,最终选择SPAdes为默认的重组装软件,这也是基于效率的考虑。事实上,Unicycler在单菌2+3代混装下的重组装效果远远好于SPAdes,但Unicycler重组装的耗时为SPAdes数倍。故在重组装过程中,我们选择了SPAdes作为默认软件,Unicycler为可选的软件。
BASALT已经可以比较好的支持纯2代数据的分箱分析及2+3的混装数据的分析,但基于纯3代测序数据组装文件的分析仍在开发中。BASALT仍有大量改进空间,我们将进一步解决3代测序数据的分析问题及效率问题。感谢用户的耐心,期望大家和我们一同提出BASALT的问题,如遇到问题,请直接和我们联系,可发邮件至yuke.sz@pku.edu.cn进行反馈,我们将陆续完善整个软件包的功能。谢谢!
(二)依赖程序
1)系统及脚本语言
Linux x64系统,Python (>=3),Perl, Java (>=1.7)。
Python附加脚本包:biopython,pandas,numpy,scikit-learn,copy,multiprocessing,collections,pytorch,tensorboardx。
2)内置的自动分箱软件
MetaBAT2,Maxbin2,CONCOCT,Semibin2,Metabinner(可选,默认不使用)。
*VAMB也存在于BASALT环境中,但内部测试的结果显示VAMB的效果并不是很好,故我们暂时阻隔了通过BASALT对该软件的调用。若想使用VAMB的用户,可以自行安装VAMB进行分箱后,使用data feeding将分箱结果输入至BASALT中。
3)映射、解析、预测及比对软件
Bowtie2,BWA,SAMtools,Prodigal,BLAST+,HMMER,Minimap2。
4)组装及碱基修正软件
SPAdes,IDBA-UD,Pilon,Unicycler(可选,默认不使用)。
5)基因组评估软件
CheckM:CheckM数据库,pplacer。
CheckM2:可选,默认不使用。BASALT中已包含了CheckM2的主程序,但BASALT环境中没有默认下载CheckM2数据库。
*由于CheckM2为基于神经网络设计的程序;我们在以标准基因组进行评估的过程中发现CheckM2与CheckM有大量的偏差,故现阶段我们仍倾向于使用CheckM而非CheckM2进行质量评估。未来在CheckM2更加成熟,或有可替代软件的情况下将会进行修正。用户如需使用CheckM2,可使用参数-qc进行选择,详见下文说明。
*BASALT环境中没有配置CheckM2数据库。用户需手动配置CheckM2的数据库(https://github.com/chklovski/CheckM2,可参考CheckM2页面)。
6)工作站硬件要求
8+内核,128GB+RAM;暂不支持显卡加速,该功能仍在开发中,将在后续版本推出。
(三) 安装方法
1)在外网或翻墙的情况下,使用BASALT_setup_international.py安装
下载BASALT_setup.py
通过该命令下载并安装软件(该过程耗时较长,请耐心等待):
python BASALT_setup.py*注:该脚本已内置了BASALT的环境配置、主程序及模型的下载及安装。
2)在内网且无法翻墙的情况下使用BASALT_setup_China_mainland.py安装
下载BASALT_setup_China_mainland.py
通过该命令下载软件(该过程耗时较长,请耐心等待):
python BASALT_setup_China_mainland.py下载神经网络训练模型:从腾讯微云下载BASALT.zip(https://share.weiyun.com/r33c2gqa)并上传至您的Linux路径,随后依次执行以下三行命令:
mv BASALT.zip ~/.cache
cd ~/.cache
unzip BASALT.zip*注:由于国内访问Github时常受限,国内用户可使用以下的手动安装方式进行BASALT的安装。
3)手动配置BASALT环境(国内用户推荐用该方式安装)
由于需要安装许多依赖项,推荐使用conda安装和管理BASALT。安装Miniconda或Anaconda后,创建一个conda环境,并在特定环境中安装BASALT。
(可选)添加镜像以提高下载速度,如国内用户可用清华的conda镜像进行安装:
site=https://mirrors.tuna.tsinghua.edu.cn/anaconda
conda config --add channels ${site}/pkgs/free/
conda config --add channels ${site}/pkgs/main/
conda config --add channels ${site}/cloud/conda-forge/
conda config --add channels ${site}/cloud/bioconda/从Github克隆BASALT仓库并配置BASALT环境
git clone https://github.com/EMBL-PKU/BASALT.git
cd BASALT
conda env create -n BASALT --file basalt_env.ymlconda create的命令耗时较长,需要耐心等待。
*注:因为github在国内访问不稳定,我们同时将环境文件:basalt_env.yml存至微云之中,国内用户可通过共享文件夹下载basalt_env.yml文件:https://share.weiyun.com/xXdRiDkl,在下载完成之后,可参考上面缩写的cd BASALT命令,及conda create命令进行操作。
授权BASALT脚本。一般来说conda BASALT环境位于conda安装的子目录envs中(例如:/home/anaconda2/envs)。
chmod -R 777 your_conda/envs/BASALT/bin/**注:命令举例:conda中BASALT环境所在地址可以使用以下方式进行查看:conda info –envs,在其中找到BASALT所在的地址,如下图:
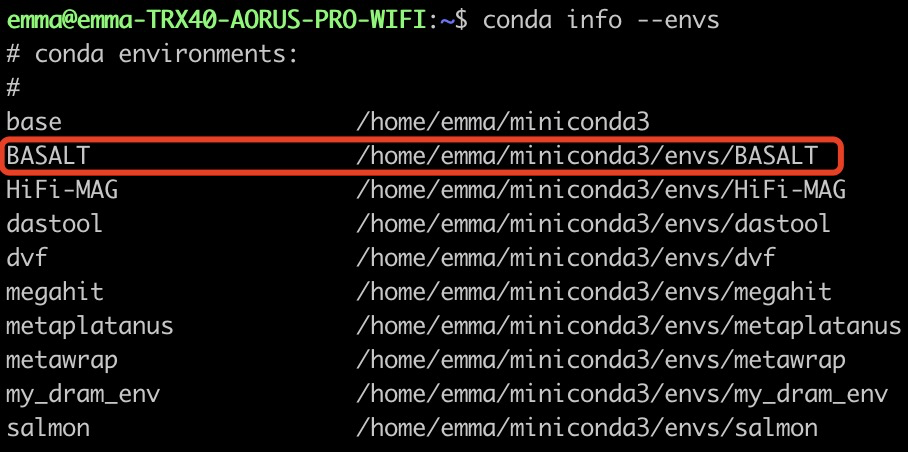
其安装的子目录为/home/emma/miniconda3/envs/BASALT,故在这一情况下使用的命令为:
chmod -R 777 /home/emma/miniconda3/envs/BASALT /bin/*下载神经网络训练模型
预先下载好BASALT_models_download.py脚本,并执行下列命令:
python BASALT_models_download.py*注:BASALT_models_download.py可通过github直接获得,为保证国内用户可以下载该文件,可访问https://share.weiyun.com/xXdRiDkl,从中下载该文件。
BASALT_models_download.py命令在国内可能会失效,国内安装推荐使用以下的方法一进行安装,国外安装也可用以下方法二进行手动安装:
方法一(内网):从腾讯微云下载BASALT.zip(https://share.weiyun.com/r33c2gqa)并上传至您的Linux路径:
mv BASALT.zip ~/.cache
cd ~/.cache
unzip BASALT.zip方法二(外网)
wget https://figshare.com/ndownloader/files/41093033
mv 41093033 BASALT.zip
mv BASALT.zip ~/.cache
cd ~/.cache
unzip BASALT.zip4)利用singularity运行预安装好的BASALT
为方便用户使用,我们提供了无需安装的BASALT singularity版本,用户只需下载预安装好的BASALT,利用singularity即可直接运行。
通过腾讯微云的网址下载BASALT.sif文件(https://share.weiyun.com/xKmoBmrF)运行命令:
singularity run BASALT.sif BASALT -h若BASALT.sif文件不在/home目录下,则需要添加-B进行挂载,命令如下:
singularity run -B /path/to/folder BASALT.sif BASALT -h运行singularity版本时,利用集群PBS等提交任务的方法可以正常运行,但直接利用nohup后台挂载运行时程序可能会意外退出。如果直接运行的话,可以使用screen方式运行,如:
screen -dmS [SESSION_NAME] bash -c 'bash BASALT.sh > BASALT.log'BASALT.sif中已安装各种需要的环境及软件,因此除运行BASALT外,还可以调用各种软件,如:
singularity run BASALT.sif bowtie2 -h
singularity run BASALT.sif checkm2 -h5)测试文件
我们准备了测试文件以测试BASALT是否成功安装并且可以顺利运行。测试文件分为三个部分:测试数据(Data.tar.gz)、用作对照看软件是否安装成功的测试结果(Final_bestbinset.tar.gz)和执行命令脚本(basalt.sh)。
下载测试文件并运行程序
从https://figshare.com/articles/dataset/BASALT_demo_files/22323424下载测试文件(共三个:Data.tar.gz、Final_bestbinset.tar.gz、basalt.sh),然后执行下面的命令运行程序(该过程耗时大约六小时,请耐心等待):
bash basalt.sh(四) 软件使用
1)参数简述
以python执行BASALT.py脚本进行操作;BASALT.py为BASALT主程序。如果使用conda环境,运行时直接输入BASALT及相关参数;如果单独使用环境,使用python BASALT.py及相关参数运行程序。以conda环境为例,若需帮助,可使用BASALT -h查看运行参数。
⭐ 必须参数
-a 输入组装文件,组间以英文逗号分隔‘,’不可出现空格。
BASALT支持用户以.fa, .fna, .fasta, .gz, .tar.gz, .zip等格式如数序列文件。
示例:-a assembly1.fa,assembly2.fa
BASALT支持单一组装文件或多组装文件进行binning;在样品的物种非常相似,但群落中物种丰度不同的情况下,如时间序列样品,多组装文件可大幅度提高binning的效果,但也可能会导致运算时间过长,但我们依然推荐大家用多组装文件模式进行分箱分析。如果运算资源不足,推荐使用多组数据进行合并组装的组装文件进行后续的分箱分析。详细可参考下文中的应用案例。
-s 输入二代测序paired-end数据,多组数据间要用‘/’间隔不同组的文件,‘/’或‘,’前后不可出现空格。
BASALT支持用户以.gz, .tar.gz, .zip等格式如数序列文件。
示例:-s d1_r1.fq,d1_r2.fq/d2_r1.fq,d2_r2.fq
BASALT支持.tar.gz等文件的输入,但是最好直接输入未压缩的序列文件。
-l 输入三代测序数据,组间以英文逗号分隔,不可出现空格。
BASALT支持用户以.gz, .tar.gz, .zip等格式如数序列文件。
示例:-l lr1.fq,lr2.fq
BASALT支持.tar.gz等文件的输入,但是最好直接输入未压缩的序列文件。
-t 输入线程数,示例:-t 32
-hf 输入HiFi数据,组间以英文逗号’,’分隔,不可出现空格。
示例:-hf hifi1.fq,hifi2.fq
-m 输入内存量,示例:-m 128,最小内存量建议设置为32G
⭐ 可选参数
-q 或--quality-check设定quality check软件的参数运行。
默认软件为CheckM,若需要使用,可用以下命令调用。
示例:-q checkm2
虽然checkm2是新出的评估软件,但我们发现checkm2与checkm在评估一些新类群时差异非常巨大,在和很多国际同行,顶尖团队的成员,包括GTDB的开发者交流之后,我们认为现阶段BASALT的默认程序仍然用CheckM为默认软件更为妥当。用户如果需要使用checkm2, 可以直接使用-q参数进行设定。
-e 设定额外分箱工具。
可选参数m,表示设定额外分箱工具为Metabinner
示例:-e m,代表BASALT将会在使用MetaBAT2,Maxbin2,CONCOCT及Semibin2作为分箱工具的同时也添加Metabinner对数据进行分箱分析。
其他工具我们仍在评估中,估计2024年7月份评估结束会更新至BASALT。
-o 或--out,设定输出文件夹的名称。
BASALT的默认输出文件夹名称为Final_bestbinset,可通过-o参数输出文件的名称。
示例:-o Human_gut,则输出文件最终的保存文件夹为 Human_gut_final_bestbinset。
-d 或--data-feeding-folder,设定额外输入的文件夹的名称。
如用户用其他binner生成了一组基因组,BASALT可通过-d参数将这组基因组整合到BASALT binning的最终的基因组中。假设用户有一个文件夹,名为Human_gut_microbiome,BASALT将自动化的读取文件夹中的基因组,将其与BASALT的得到的基因组进行去冗余、选择最优基因组并进行修正,进而获得最终的精修基因组。
示例:-d Metawrap_bins
-d参数除了可以支持BASALT跑出的基因组直接和-d提供的基因组进行融合。同时也支持用户对自己之前已经跑完的基因组利用BASALT进行修正。用户可以在将其他的基因组提供给BASALT之后,利用-b参数对多个基因组文件夹进行去冗余;或利用-r参数对这一文件夹内的基因组进行修正。详见下文示例。
-b 或--binsets-list,利用BASALT对多个binsets进行基因组的去冗余。
使用时需要将多个基因组文件夹输入给BASALT。-b参数需输入至少两个含有基因组的文件夹
示例:-b Metawrap_bins,DAStools_binset
-b参数功能类似于drep。相比于drep,BASALT在近缘样品的去冗余上有统治性的优势。具体使用时,先需要将基因组利用BASALT -d参数将基因组文件做成BASALT所需要的文件夹,之后再用-b文件去冗余。详见下文示例。
-r 或--refinement-binset,利用BASALT对多个binsets进行修正。
使用时需要将多个基因组文件夹输入给BASALT。-r参数只可输入单一文件夹。
示例:-r refinement_binset
-r参数功能用于包括BASALT自主产生或外源产生的基因组的修正工作。使用前先需要用-d参数将这些基因组文件提交给BASALT进行基因组的feeding,而后输入特定文件进行修正工作。详见下文示例。
--sensitive 基因获取及修正的敏感程度,包含三个模式:quick/sensitive/more-sensitive。
示例:--sensitive more-sensitive
现阶段quick模块包含的binner为metabat2及semibin2;sensitive模式含有metabat2、semibin2及concoct;more-sensitive模式中含有metabat2、semibin2、concoct及maxbin2,并且在more-sensitive下的refinement功能模块中含有一个深度修正的模块。虽然more-sensitive模式可以得到最多且质量最高的基因组,但因样品的差异我们很难确定和sensitive模式相比,more-sensitive模式是否更好。在我们的测试里,在more-sensitive模式下,我们的数据可以获得更多的稀有古菌的基因组,但这存在很强的随机性,更多的时候,sensitive模式与more-sensitive模式获得的基因组数量差别不大。但sensitive模式在复杂样品的情况下,耗时仅为more-sensitive模式不足一半,可大幅度的提高速度。近期我们将对软件,包括各种binner进行评测并整合至BASALT之中,预计在2024年7月的版本会进行一次较大的更新。现阶段,BASALT的默认为sensitive模式。
--module 设定BASALT的工作模块。
BASALT可单独进行autobinning + bin selection,refinement及这三个部分的独立运行,也可以以all进行完整的自动化binning,默认模式为all。
示例:--module autobinning
--min-cpn 设定保留的基因组的最小完整度,默认值为35,示例:--min-cpn 30。
--max-ctn 设定保留的基因组的最大污染度,默认值为20,示例:--max-ctn 25。
--mode 设定运行模式,预设new及continue两种模式。New模式为重新开始binning项目,continue为断点续跑跑上一次任务未完成的任务,默认模式为continue。
--refinepara 设定refinement模块的运行参数,预设deep和quick两个参数,deep参数可将基因组修正的更好,但会相对耗费更多时间,默认参数为deep。
-h 帮助文档
2)使用范例
1. 单独使用二代测序数据进行binning及修正
BASALT -a as1.fa,as2.fa,as3.fa -s ds1_r1.fq,d1_r2.fq/d2_r1.fq,d2_r2.fq -t 60 -m 2502. 使用二代测序数据及三代测序数据进行binning及修正
BASALT -a as1.fa,as2.fa,as3.fa -s ds1_r1.fq,d1_r2.fq/d2_r1.fq,d2_r2.fq -l lr1.fq,lr2.fq -t 60 -m 2503. 使用hifi数据进行binning及修正
BASALT -a as1.fa -hf hifi1.fq -t 60 -m 2504. 使用hifi数据及二代测序数据进行binning及修正
BASALT -a as1.fa -hf hifi1.fq -s ds1_r1.fq,d1_r2.fq -t 60 -m 2505. 在BASALT使用checkm2
BASALT -a as1.fa,as2.fa,as3.fa -s ds1_r1.fq,d1_r2.fq/d2_r1.fq,d2_r2.fq -t 60 -m 250 -q checkm26. 使用BASALT进行基因组的获取,并且融合之前已有的基因组数据
BASALT -a as1.fa,as2.fa,as3.fa -s ds1_r1.fq,d1_r2.fq/d2_r1.fq,d2_r2.fq -d m_binset -t 60 -m 2507. 使用BASALT进行外源数据基因组的去冗余
a) 数据输入(Data feeding)
BASALT -s sample1.R1.fq, sample1.R2.fq/sample2.R1.fq, sample2.R2.fq -d mbin,dbin -t 60 -m 250-d参数会产生一个Date_feeded文件夹,其中含有BASALT做后续工作需要的修改了index的组装文件,如下文中的500_mbin.fa;基因组文件夹,如500_mbin.fa_BestBinsSet;覆盖率文件,如Coverage_matrix_for_binning_500_mbin.fa.txt;及修改过格式的reads文件。
b) 如冗余(De-replication)
填入BASALT所需的多组基因组文件夹、多个组装文件、多对reads及覆盖率文件。该过程将产生一个去冗余后的基因组文件夹BestBinset。
BASALT -b 500_mbin.fa_BestBinsSet,501_dbin.fa_BestBinsSet -c Coverage_matrix_for_binning_500_mbin.fa.txt, Coverage_matrix_for_binning_501_mbin.fa.txt -a 500_mbin.fa,501_mbin.fa -s sample1.R1.fq, sample1.R2.fq/ sample2.R1.fq, sample2.R2.fq -t 608. 使用BASALT进行基因组的修正
在数据输入之后(data feeding),或者去冗余完成后,将基因组文件通过-r参数输入到BASALT内。完成后脚本会生成BestBinset_outlier_refined文件夹。
BASALT -r BestBinset -c Coverage_matrix_for_binning_500_mbin.fa.txt, Coverage_matrix_for_binning_501_mbin.fa.txt -a 500_mbin.fa,501_mbin.fa -s PE_r1_sample1.R1.fq,PE_r2_sample1.R2.fq/PE_r1_sample2.R1.fq, PE_r2_sample2.R2.fq -t 603)使用举例
项目一:水产养殖肠道样本组装及分箱策略
项目背景
采集三种不同水产养殖生物池塘中:养殖生物肠道、养殖水环境、养殖塘沉积物样本共计27份,进行宏基因组二代测序及三代测序。
组装策略
首先,通过OPERA-MS软件对二代测序数据及三代测序数据进行单个样本的混合组装;同时,对所获取的相同类型样品平行样的双端数据及三代测序数据合并后进行多个样本的混合组装,以获取该类型样本更多维度信息,便于后续分箱处理;在获取单个样本及多个平行样本混合的组装文件后,进行分箱处理。
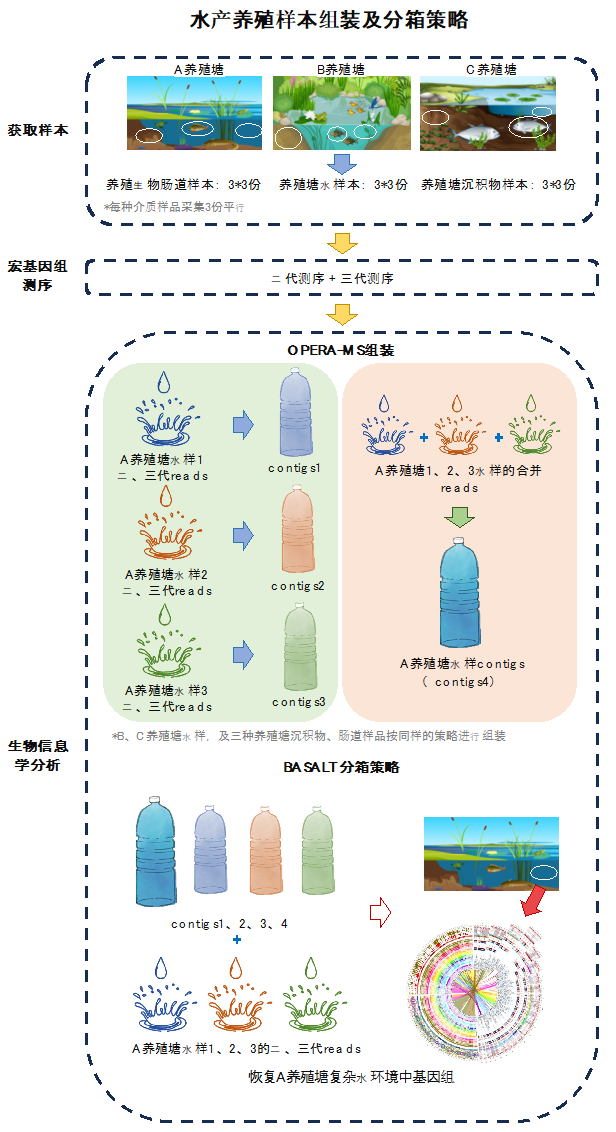
图1 水产养殖样本组装及分箱策略
样本数据(以A养殖塘水环境样本为例)
sampleA_w1_R1.fq, sampleA_w1_R2.fq,
sampleA_w2_R1.fq, sampleA_w2_R2.fq,
sampleA_w3_R1.fq, sampleA_w3_R2.fq
sampleA_w1_lr.fq
sampleA_w2_lr.fq
sampleA_w3_lr.fq组装结果
sampleA_w1_assembly.fa
sampleA_w2_assembly.fa
sampleA_w3_assembly.fa
sampleA_w1+A_w2+A_w3_assembly.fa分箱命令
BASALT \
-a sampleA_w1_assembly.fa,sampleA_w2_assembly.fa,sampleA_w3_assembly.fa,sampleA_w1+A_w2+A_w3_assembly.fa \
-s sampleA_w1_R1.fq,sampleA_w1_R2.fq/sampleA_w2_R1.fq,sampleA_w2_R2.fq/sampleA_w3_R1.fq,sampleA_w3_R2.fq \
-l sampleA_w1_lr.fq,sampleA_w2_lr.fq,sampleA_w3_lr.fq \
-t 60 -m 250项目二:不同生长周期鸟类肠道样本组装及分箱策略
项目背景
在鸟类生长不同时期采集若干种不同鸟类粪便样本,进行宏基因组二代测序。
组装策略
首先,通过metaspades软件对二代测序数据进行单个样本的组装;同时,对所获取的同种鸟类的同一生长周期样品平行样的双端数据数据合并后进行多个样本的组装,以获取该生长周期样本更多维度信息,便于后续分箱处理;在获取单个样本及多个平行样本混合的组装文件后,进行分箱处理。
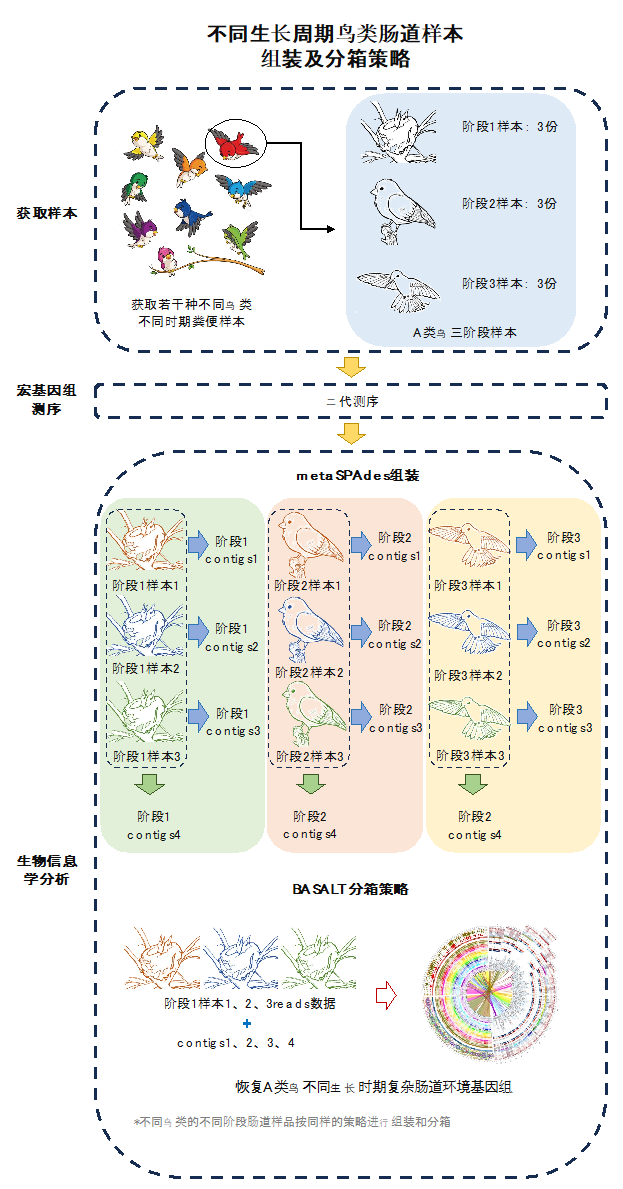
图2 不同生长周期鸟类肠道样本组装及分箱策略
样本数据(以A鸟样本为例)
(A类鸟阶段1样本)
sampleA_s1_1_R1.fq, sampleA_s1_1_R2.fq,
sampleA_s1_2_R1.fq, sampleA_s1_2_R2.fq,
sampleA_s1_3_R1.fq, sampleA_s1_3_R2.fq(A类鸟阶段2样本)
sampleA_s2_1_R1.fq, sampleA_s2_1_R2.fq,
sampleA_s2_2_R1.fq, sampleA_s2_2_R2.fq,
sampleA_s2_3_R1.fq, sampleA_s2_3_R2.fq(A类鸟阶段3样本)
sampleA_s3_1_R1.fq, sampleA_s3_1_R2.fq,
sampleA_s3_2_R1.fq, sampleA_s3_2_R2.fq,
sampleA_s3_3_R1.fq, sampleA_s3_3_R2.fq组装结果:
sampleA_s1_1_assembly.fa,sampleA_s1_2_assembly.fa,sampleA_s1_3_assembly.fa
sampleA_s2_1_assembly.fa,sampleA_s2_2_assembly.fa,sampleA_s2_3_assembly.fa
sampleA_s3_1_assembly.fa,sampleA_s3_2_assembly.fa,sampleA_s3_3_assembly.fasampleA_s1_assembly.fa
sampleA_s2_assembly.fa
sampleA_s3_assembly.fa分箱命令(一阶段样品分箱命令)
BASALT -a sampleA_s1_assembly.fa, sampleA_s1_1_assembly.fa,sampleA_s1_2_assembly.fa,sampleA_s1_3_assembly.fa \
-s sampleA_s1_1_R1.fq,sampleA_s1_1_R2.fq/sampleA_s1_2_R1.fq,sampleA_s1_2_R2.fq/sampleA_s1_3_R1.fq,sampleA_s1_3_R2.fq \
-t 60 -m 250*注:二、三阶段样品分箱命令和一阶段相同,仅需替换对应文件即可
(五) 安装和运行中常见问题
1)按照官网流程安装后在运行BASALT的过程中遇到报错
samtools: error while loading shared libraries: libcrypto.so1.0.0: cannot open shared object file: No such file or directory解决方案:
a) 首先查看安装BASALT环境的路径下是否存在libcrypto.so1.0.0:
~/anaconda3///BASALT/lib$ ls libcry*b) BASALT环境路径下如果存在libcrypto.so.1.1但是不存在libcrypto.so.1.0.0则建立软链接将libcrypto.so.1.1链接到libcrypto.so.1.0.0:
~/anaconda3/<path>/<to>/BASALT/lib$ ln -s ~/anaconda3/<path>/<to>/BASALT/lib/libcrypto.so.1.1 ~/anaconda3/<path>/<to>/BASALT/lib/libcrypto.so.1.0.0c) 在BASALT环境下测试samtools能否使用:
samtools –help2)运行BASALT的过程中遇到报错1
Traceback (most recent call last):
File "/users/raw937/.conda/envs/BASALT/bin/BASALT", line 57, in
datasets[str(n)].append(pr[1].strip())
IndexError: list index out of range解决方案:该问题是由于BASALT不支持使用路径输入序列文件导致,可以通过建立一个单独的文件夹用于存放需要处理的文件来解决。运行命令可以参考:
BASALT -a a1.fa,a2.fa -s seq1_r1.fq,seq1_r2.fq/seq2_r1.fq,seq2_r2.fq -l ont_seq.fq -t 60 -m 2503)运行BASALT的过程中遇到报错2
Traceback (most recent call last):
File "/dss/dsshome1/lxc04/ge24yaf2/.conda/envs/BASALT/bin/BASALT", line 53, in
datasets_list=sr_datasets.split('/')解决方案:该问题是由于BASALT不支持仅输入三代测序文件导致(目前版本仅支持PacBio-HiFi的纯三代数据输入),可以通过输入二代+三代测序文件来解决。
4)运行BASALT的过程中遇到报错3
INFO: Running CheckM2 version 1.0.1
[03/13/2024 12:56:34 PM] INFO: Running quality prediction workflow with 30 threads.
[03/13/2024 12:56:34 PM] ERROR: DIAMOND database not found. Please download database using <checkm2 database --download>解决方案:该问题是由于CheckM2数据库无法在conda安装过程中自动安装导致,可以通过按照CheckM2安装指南手动下载来解决。CheckM2安装指南:https://github.com/chklovski/CheckM2。
5)运行BASALT的过程中遇到报错4
BASALT: command not found解决方案:将BASALT_script.zip放入BASALT环境下(一般路径为/home/user/anaconda2/envs)。
unzip BASALT_script.zip
chmod -R 777 BASALT_scrip
mv BASALT_scrip /* your_conda/envs/BASALT/bin6)运行BASALT的过程中遇到报错5
Traceback (most recent call last):
File "/home/pthierin/miniconda3/envs/BASALT/bin/BASALT", line 137, in <module>
BASALT_main_d(assembly_list, datasets, num_threads, lr_list, hifi_list, hic_list, eb_list, ram, continue_mode, functional_module, autobining_parameters, refinement_paramter, max_ctn, min_cpn, pwd, QC_software)
File "/home/pthierin/miniconda3/envs/BASALT/bin/BASALT_main_d.py", line 494, in BASALT_main_d
Contig_recruiter_main(best_binset_from_multi_assemblies, outlier_remover_folder, num_threads, continue_mode, min_cpn, max_ctn, assembly_mo_list, connections_list, lr_connection_list, coverage_matrix_list, refinement_paramter, pwd)
File "/home/pthierin/miniconda3/envs/BASALT/bin/S6_retrieve_contigs_from_PE_contigs_10302023.py", line 1819, in Contig_recruiter_main
parse_bin_in_bestbinset(assemblies_list, binset+'_filtrated', outlier_remover_folder, PE_connections_list, lr_connection_list, num_threads, last_step, coverage_matrix_list, refinement_mode)
File "/home/pthierin/miniconda3/envs/BASALT/bin/S6_retrieve_contigs_from_PE_contigs_10302023.py", line 1695, in parse_bin_in_bestbinset
bin_comparison(str(binset), bins_checkm, str(binset)+'_retrieved', refinement_mode, num_threads)
File "/home/pthierin/miniconda3/envs/BASALT/bin/S6_retrieve_contigs_from_PE_contigs_10302023.py", line 731, in bin_comparison
for line in open('quality_report.tsv','r'):
FileNotFoundError: [Errno 2] No such file or directory: 'quality_report.tsv'解决方案:该问题是由于数据coverage较低导致binning文件夹中的bins数量过少,从而导致quality_report.tsv无法生成。
BASALT -a a1.fa,a2.fa -s seq1_r1.fq,seq1_r2.fq/seq2_r1.fq,seq2_r2.fq -l ont_seq.fq -t 60 -m 250(六) 常见其他问题(FAQ)
Q1:在单独组装与合并组装文件同时输入的情况下,某些contigs被重复利用,会影响分箱结果吗?
A1:在单独组装与合并组装文件同时输入的情况下,确实会产生冗余的基因组,如:bin1从sampleA1_assembly.fa中获得,bin2从sampleA1A2A3_assembly.fa中获得,而bin1与bin2实际上为同一个基因组。考虑到这种情况的产生,BASALT在bin selection中可以识别并去除冗余的基因组,因此最终产出的基因组为非冗余基因组。
Q2:与metaWRAP相比,BASALT运行速度通常会更慢。是否有加快运行的方法?
A2:在使用单个组装文件运行的过程中,BASALT的完整流程所耗时间确实会比metaWRAP要多出一倍左右,其耗时比例还会随着样品的复杂度增加而进一步增加。但是,在文章中也提到,即使不运行gap filling模块,BASALT所得到的基因组质量与数量均比metaWRAP要好。实际上,在多个组装文件输入模式下,BASALT比metaWRAP会更省时,因为BASALT只需运行一次,且其单独组装+合并组装+去冗余的模式可以大大增加非冗余基因组的产出。如用户对基因组的深度挖掘要求不高,且需要加快整个分箱进程,建议以下两种策略:
(1) 使用MetaBAT2 + Semibin2作为初始Binners,可以大大加快auto-binning的进程;
(2) 仅运行BASALT中的auto-binning + bin selection + refinement模块,不使用gap filling模块可以减少运行时间;
(3) 在多个样品同时输入的情况下,仅使用单一的合并组装文件(如:sampleA1A2A3_assembly.fa),可以减少auto-binning的运算时间,但也会相应减少基因组的产生。
*注:BASALT的三种运行模式仍在测试之中,预计在BASALT-1.0.2版本中会得到更新(约2024年5月更新),请耐心等待。
Q3:我已经有之前分箱得到的基因组,想要直接进行refinement,该如何操作?
A3:在已有基因组的情况下,可以利用data feeding模块进行数据的输入。
*注:BASALT的data feeding运行模式仍在测试之中,预计在BASALT-1.0.2版本中会得到更新(约2024年5月更新),请耐心等待。
Q4:运行BASALT的过程中我需要设定输出路径吗?
A3:不需要。BASALT在输出时候自动将结果及重要过程文件输出至当前工作路径下,建议用户将需要分析的文件单独建立文件夹存放和运行BASALT,以免重复输出覆盖原有结果。
(七) 更新日志
BASALT v1.1.0:
更改了module mode的默认模式为‘sensitive’;
更改了refinement的默认模式为‘quick’;
将quality check的参数名称‘-qc’改为‘-q’;
增加了‘-o’参数,对最终输出的final binset文件夹进行重命名;
增加了data feeding的模式:-d, -b, 和-r。现在用户可以直接输入利用其他软件获得的bins作为输入文件,对这些bins进行去冗余和进一步修正。
修正了一些错误。
(八) 软件引用
Z Qiu, L Yuan, C Lian, B Lin, J Chen, R Mu, X Qiao, L Zhang, Z Xu, L Fan, Y Zhang, S Wang, J Li, H Cao, B Li, B Chen, C Song, Y Liu, L Shi, Y Tian, J Ni, T Zhang, J Zhou, W Zhuang, K Yu. BASALT refines binning from metagenomic data and increases resolution of genome-resolved metagenomic analysis. Nature Communications 2024, 15, 2179. https://doi.org/10.1038/s41467-024-46539-7
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 2238
2238

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









