






易扩增子:易用、可重复和跨平台的扩增子分析流程
EasyAmplicon: An Easy-to-use, Reproducible and Cross-platform Pipeline for Amplicon Analysis
刘永鑫1, 2, 3, #, *,陈同4, #,周欣5,白洋1, 2, 3, 6, *
1中国科学院遗传与发育生物学研究所,植物基因组学国家重点实验室,北京;2中国科学院大学,生物互作卓越创新中心,北京;3中国科学院遗传与发育生物学研究所,中国科学院–英国约翰英纳斯中心植物和微生物科学联合研究中心,北京;4中国中医科学院,中药资源中心,北京;5中国科学院微生物研究所,真菌学国家重点实验室,北京;6中国科学院大学现代农学院,北京
#共同第一作者
引文:Yong-Xin Liu, Lei Chen, Tengfei Ma, Xiaofang Li, Maosheng Zheng, Xin Zhou, Liang Chen, Xubo Qian, Jiao Xi, Hongye Lu, Huiluo Cao, Xiaoya Ma, Bian Bian, Pengfan Zhang, Jiqiu Wu, Ren-You Gan, Baolei Jia, Linyang Sun, Zhicheng Ju, Yunyun Gao, Tao Wen, Tong Chen. 2023. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2: e83. https://doi.org/10.1002/imt2.83
摘要:扩增子测序是目前微生物组研究中最广泛的使用手段,主流的分析流程有QIIME、USEARCH和Mothur,但仍分别存在依赖关系过多导致的安装困难、大数据收费和使用界面不友好等问题。本文搭建了一套完整的扩增子分析流程命名为易扩增子(EasyAmplicon),可以实现简单易用、可重复和跨平台地开展扩增子分析。流程计算核心采用体积小、安装方便、计算速度快且跨平台的软件USEARCH,同时整合VSEARCH以突破USEARCH免费版的限制。分析选用RStudio的图形界面对流程代码文档管理和运行,实现命令行和/或鼠标点击操作方式开展扩增子可重复分析。同时流程还提供了数10个脚本,实现特征表过滤、重采样、分组均值等常计算,以及为常用软件如STAMP、LEfSe、PICRUSt1/2等提供标准的输入文件的功能。易扩增子可以从https://github.com/YongxinLiu/EasyAmplicon下载轻松部署于Windows/Mac/Linux操作系统,在普通个人电脑上2小时内可完成数十个样本的分析。
关键词:扩增子,分析流程,16S,ITS,USEARCH
仪器设备
1.个人电脑/服务器 (操作系统:Windows 10/Mac OS 10.12+/Linux Ubuntu 18.04+;CPU:2核+;内存:8G+;硬盘:> 10 GB,且大于10倍原始数据大小),网络访问畅通。
软件和数据库
1.R语言环境,下载适合自己系统的4.0.2版:https://www.r-project.org/
2.R语言开发环境,用于执行流程,下载适合自己系统的RStudio 1.3.1056:https://www.rstudio.com/products/rstudio/download/#download
3.(可选) 仅Windows系统安装,提供Git Bash命令行环境的GitForWidnows 2.28.0:http://gitforwindows.org/
4.扩增子分析流程USEARCH v10.0.240 (Edgar, 2010) https://www.drive5.com/usearch/download.html
5.扩增子分析流程VSEARCH v2.15.0 (Rognes等,2016) https://github.com/torognes/vsearch/releases
6.易扩增子流程EasyAmplicon v1.10 (Zhang等,2018和2019; Chen等,2019; Huang等,2019; Liu等,2020; Qian等,2020a和2020b):https://github.com/YongxinLiu/EasyAmplicon
7.核糖体数据库RDP v16 (Cole等,2014):http://rdp.cme.msu.edu/
8.(可选) 核糖体数据库GreenGene数据库(gg) 13_8(McDonald等,2011):ftp://greengenes.microbio.me/greengenes_release
9.(可选) 核糖体数据库SILVA v123 (Quast等,2013):http://www.arb-silva.de
10.(可选) 转录间隔区(ITS)数据库UNITE v8.2(Nilsson等,2019):https://unite.ut.ee/
11.(可选) Windows版下载工具wget:http://gnuwin32.sourceforge.net/packages/wget.htm
软件安装和数据库部署
注:以下的软件安装和使用均在64位Windows 10系统中演示,Linux/Mac中不同的地方会有说明,流程代码提供有Mac版本(pipeline_mac.sh)。
Windows系统需要安装GitForWindows (http://gitforwindows.org/) 提供Git bash环境支持常用Shell命令。Linux/Mac系统自带Bash命令行工作环境。
以64位Windows 10系统为例,我们先安装R/RStudio软件,再把本流程 (EasyAmplicon/目录)保存于C盘中,然后根据需要下载数据库至指定目录即完成部署。
注:代码行添加灰色底纹背景,其中需要根据系统环境修改的部分标为蓝色。
1.流程运行环境R和RStudio
依次安装适合系统的最新版R语言 (https://www.r-project.org) 和RStudio (https://www.rstudio.com/products/rstudio/download/)。注意操作系统用户名不要使用中文,否则会影响R语言使用。
2.批量安装依赖R包
流程会调用数百个R包,使用时可自动安装。但由于网络或系统等个性原因经常出现下载或安装失败,可以使用中根据提示手动安装缺失R包。本文推荐直接下载我们预编译好的R包合辑 (http://nmdc.cn/datadownload),替换至R包所在目录即可,详见常见问题1。
3.易扩增子流程EasyAmplicon
访问https://github.com/YongxinLiu/EasyAmplicon,选择Code—Download ZIP下载并解压,如保存于C盘并确保目录名为EasyAmplicon。如在RStudio的Terminal中可使用git下载流程:
git clone git@github.com:YongxinLiu/EasyAmplicon.git
4.(可选) 扩增子流程依赖软件
EasyAmplicon依赖的Windows/Mac/Linux版本软件已经保存于EasyAmplicon中的win/mac/linux目录中,如果出现问题,可按如下方法手动安装。
USEARCH下载页https://www.drive5.com/usearch/download.html,选择适合自己系统的10.0.240版本 (不要下载最新版,因为有更多功能使用受限),如Windows版本保存至EasyAmplicon目录中的win目录中,解压后改名为usearch.exe。Linux/Mac系统需下载到环境linux/mac目录,解压后改名为usearch,并添加可执行权限(chmod +x usearch)。VSEARCH下载页面https://github.com/torognes/vsearch/releases,选择适合自己系统的最新版下载,接下来操作与USEARCH类似。Windows系统还需下载wget程序(http://gnuwin32.sourceforge.net/packages/wget.htm) 至win目录。
5.(可选)16S核糖体基因物种注释数据库
16S扩增子测序分析,常用RDP/SILVA/GreenGene数据库进行物种注释,可以从上述数据库官网下载并整理为USEARCH使用的格式,此处推荐从USEARCH官网 (http://www.drive5.com/sintax) 下载USEARCH兼容格式的数据库。默认流程使用体积小巧的RDP v16数据库(rdp_16s_v16_sp.fa.gz),并已保存于usearch目录中。可选GreenGenes 13.5(gg_16s_13.5.fa.gz)和SILVA(silva_16s_v123.fa.gz)数据库,可根据需要下载并保存于usearch目录中。此外,如果要开展PICRUSt和Bugbase功能预测分析,还需要使用GreenGenes数据库13.5中按97%聚类的OTU序列(己保存于流程gg目录中97_otus.fasta.gz)。可选手动下载GreenGenes官方数据库(ftp://greengenes.microbio.me/greengenes_release),解压后选择其中的97_otus.fasta保存于gg目录下即可。
6.(可选)ITS物种注释数据库
如果研究真菌或真核生物采用转录间隔区(Intergenic Transcribed Spacer)测序,需要使用UNITE数据库,目前最新版已经保存于usearch目录(utax_reference_dataset_all_04.02.2020.fasta.gz)。如流程中数据库没有及时更新,可在UNITE官网(https://unite.ut.ee/)下载最新版适合USEARCH的注释数据库。官方数据库存在格式问题,详细常见问题2。
实验步骤
开始新项目分析前,我们需要在项目目录(如c:/test)中准备三类起始文件:1. EasyAmplicon流程中复制分析流程文件(pipeline.sh);2. 编写样本元数据(metadata.txt);3. seq目录存放测序数据(*.fq.gz)。然后使用RStudio打开pipeline.sh,设置分析流程(EasyAmplicon, ea)和工作目录(work directory, wd)位置,添加依赖可执行程序至环境变量,并切换至工作目录。
注:用户请根据操作系统类型、软件和工作目录实际位置自行修改。
ea=/c/EasyAmplicon
wd=/c/test
PATH=$PATH:${ea}/win
cd ${wd}
1.准备输入数据 (测试数据是流程正对照)
建议下载测试元数据和测序数据作为实验的正对照,首先完成全部流程分析,以确定流程部署成功。将来使用自己的数据出现问题,可以与测试数据分析中对应结果比较,以便确定问题产生的原因。
下载示例元数据用于参考编写格式(表1)。
wget -c http://210.75.224.110/github/EasyAmplicon/data/metadata.txt
表1.元数据格式示例
SampleID | Group | Date | Site | CRA | CRR |
KO1 | KO | 2017/6/30 | Chaoyang | CRA002352 | CRR117575 |
KO2 | KO | 2017/6/30 | Chaoyang | CRA002352 | CRR117576 |
KO3 | KO | 2017/7/2 | Changping | CRA002352 | CRR117577 |
可用Excel编写,保存存为制表符分隔的的文本文件。
注意:有行列标题,行为样品名(字母开头+数字组合),列为分组信息(至少1列,可多列)、地点和时间(提交数据必须)、及其它属性,详见附表1,或下载的metadatat.txt文件。
通常测序公司会返回原始数据,如Illumina双端测序的文件,每个样本有一对文件。本文使用的数据来自发表于Science杂志关于拟南芥根系微生物组研究的文章(Huang等,2019),GSA项目号为PRJCA001296。为方便演示流程的使用,我们从中选取三个组(每组包括6个生物学重复共18个样本),并且随机抽取了50000对序列作为软件的测序数据,该数据可以从中国科学院基因组研究所的原始数据归档库 (Genome Sequence Archive,GSA,https://bigd.big.ac.cn/gsa/) (Wang等,2017)中按批次编号CRA002352搜索并下载。本文使用wget根据样本元数据中批次和样本编号批量下载至seq目录,代码如下。
mkdir -p seq
awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_f1.fq.gz -O seq/"$1"_1.fq.gz")}' <(tail -n+2 metadata.txt)
awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_r2.fq.gz -O seq/"$1"_2.fq.gz")}' <(tail -n+2 metadata.txt)
awk为Linux下的一种字符处理语言,可同时使用文本中的多个字段;使用system命令调用wget,实现根据列表批量下载、改名的功能。
检查文件大小,确定是否下载完整或正常。
ls -lsh seq
数据库第一次使用需要解压
gunzip ${ea}/usearch/rdp_16s_v16_sp.fa.gz
gunzip ${ea}/gg/97_otus.fasta.gz
创建临时和结果目录,临时目录分析结束可删除
mkdir -p temp result
2.合并双端序列并按样品重命名
依照实验设计采用for循环批处理样本合并(图1)。tail -n+2去表头,cut -f 1取第一列,即获得样本列表。vsearch程序的--fastq_mergepairs命令实现双端序列合并,接输入读长文件1(fq/fq.gz均可),--reverse接读长文件2,--fastqout指定输出文件,--relabel将序列按样本名进行重命名 (注:样本名后必须添加点,以分隔样本名和序列ID,同时注意样本名中不允许有点)。本示例数据包括5万对读长的18个样本合并计算耗时约2分钟。Windows下复制命令Ctrl+C为Linux下的终止命令,为防止异常中断,结尾添加&使命令转后台。注:如分析时提示文件质量值问题,详见常见问题3;如输入文件为单端FASTQ文件,则只需序列改名即可,详见常见问题4。
for i in `tail -n+2 result/metadata.txt | cut -f 1`;do
vsearch --fastq_mergepairs seq/${i}_1.fq.gz --reverse seq/${i}_2.fq.gz \
--fastqout temp/${i}.merged.fq --relabel ${i}.
done &
双端合并且重命名的序列,每条序列具有唯一且可识别样本的ID。可以合并所有样品至同一文件,方便统一操作。
cat temp/*.merged.fq > temp/all.fq
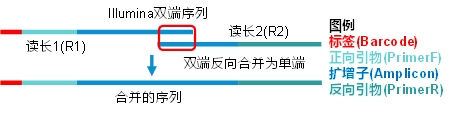
图1. 典型扩增子测序双端序列合并结构图
3.引物切除和质量控制
采用等长的方式切除引物,引物外侧如果有标签(Barcode),标签的长度需要计算在内(图1)。如本示例的结构为10 bp左端标签 + 19 bp正向引物V5共计29 bp,18 bp反向引物V7,分别使用--fastq_stripleft和--fastq_stripright传递给程序。注意:务必清楚实验设计中引物和标签长度,如果引物已经去除,可在下方参数处填0表示无需去除。此外,--fastq_maxee_rate指定质量控制的错误率,0.01代表要求错误率小于1%。质控控制后,--fastaout输出体积更小的无质量值fasta(fa)格式文件。
vsearch --fastx_filter temp/all.fq \
--fastq_stripleft 29 --fastq_stripright 18 \
--fastq_maxee_rate 0.01 \
--fastaout temp/filtered.fa
4.序列去冗余并挑选代表序列(OTU/ASV)
序列去冗余可以使总数据量降低数量级(降维),减少下游计算资源消耗和缩短等待时间,更重要的是提供序列的出现频次对鉴定真实特征序列至关重要。扩增子分析中特征序列有可操作分类单元(OTUs)或扩增序列变体(ASVs)。参数--miniuniqusize设置使用序列的最小出现频次,默认为8,此处设置为10,推荐最小值为总数据量的百万分之一(如1亿条序列至少需要过滤掉频次100以下的序列噪音),可实现去除低丰度或测序噪音并极大地增加计算速度。-sizeout输出频次, --relabel设置输出序列前缀(示例输出序列ID:>Uni1;size=17963)。
vsearch --derep_fulllength temp/filtered.fa \
--output temp/uniques.fa \
--relabel Uni --minuniquesize 10 --sizeout
按97%聚类生成OTUs和去噪挑选ASVs是目前挑选特征序列的两种主流方法。通常两种分析方法的结果整体上比较类似,细节上会略有不同。下面按方法1或2分别介绍,用户可根据实际需求选择。如聚类或去噪运行中提示超过内存限制错误,表明数据低丰度和/或测序噪音较多,可增加上步--minuniquesize参数(如30,50或更大),使输出非冗余序列数据小于1万条为宜,保证下游分析顺利且高效完成。
方法1. 使用UPARSE(Edgar,2013)算法按97%的序列相似度聚类OTU
usearch -cluster_otus temp/uniques.fa \
-otus temp/otus.fa \
-relabel OTU_
此方法累计使用次数最多、分析速度快,适合大数据或ASV方法结果规律不明显时尝试。
方法2. 使用UNOISE算法 (Edgar和Flyvbjerg,2015) 去噪生成ASV
此方法是当前的主流,推荐优先使用。类似于按100%的序列相似度聚类,或不聚类的方法,详见方法原始文献(Edgar和Flyvbjerg,2015)或宏基因组公众号推文《扩增子分析还聚OTU就真OUT了》。采用更先进的方法来鉴定测序过程中可能的错误,因此也需要消耗更多的计算时间。UNOISE算法虽然慢于UPARSE算法,但也比同类去噪算法Deblur和DADA2分别快10倍和100倍 (Amir等,2017)。此处-unoise3去噪结果默认前缀为Zotu,我们修改为主流使用的ASV。
usearch -unoise3 temp/uniques.fa \
-zotus temp/zotus.fa
sed 's/Zotu/ASV_/g' temp/zotus.fa > temp/otus.fa
(可选) 基于参考去嵌合。
全头(de novo)去嵌合时要求亲本丰度为嵌合体16倍以上防止造成假阴性,而参考数据库无丰度信息,只需1条序列在参考数据中没有相似序列且与2~3条序列相似即判定为嵌体,因此容易引起假阴性(真实序列被当作假序列丢弃),不推荐使用。如果必须要使用,由于已知序列不会被去除,选择越完整的数据库可降低假阴性率。
方法1. VSEARCH结合RDP数据库去嵌合(快但容易假阴性),推荐SILVA去嵌合(silva_16s_v123.fa),但计算极耗时 (本例用时3小时,是RDP计算时间的30倍以上)。
vsearch --uchime_ref temp/otus.fa \
-db ${ea}/usearch/rdp_16s_v16_sp.fa \
--nonchimeras result/raw/otus.fa
Windows系统下vsearch结果会添加了windows换行符^M必需删除,否则会出现换行混乱的问题。Mac/Linux系统无须执行此命令。
sed -i 's/\r//g' result/raw/otus.fa
方法2. 不去嵌合。
cp -f temp/otus.fa result/raw/otus.fa
5.特征表生成和筛选
5.1生成特征表
使用vsearch的--usearch_global命令比对扩增子序列(temp/filtered.fa)至特征序列(result/raw/otus.fa)即可生成特征表,--threads设置整数使用计算机可用的多线程加速计算。
vsearch --usearch_global temp/filtered.fa --db result/raw/otus.fa \
--otutabout result/raw/otutab.txt --id 0.97 --threads 4
sed -i 's/\r//' result/raw/otutab.txt
5.2按物种注释筛选特征表
基于物种注释,(可选)可以筛选质体和非细菌和非古菌去除并统计比例。
使用USEARCH的sintax算法进行物种注释。选择RDP物种注释(rdp_16s_v16_sp)数据库具有体积小、分类速度极快(本例耗时15秒)的特点,但缺少真核数据无法注释真核污染物来源详细。SILVA数据库(silva_16s_v123.fa)可以更好地注释真核质体序列来源,但速度较慢(本例耗时约3小时);还可选Greengenes数据库(gg_16s_13.5.fa),但此数据库自13年起再无更新。--sintax_cutoff设置分类结果的可信度阈值,范围0~1之间,文章中常用0.6/0.8,取值越大注释结果越可靠同时注释比例也越低。注意,结果中第三列方向正常应全为正向(+),如果全为反向(-),请参考常见问题5中方法将第3步结果序列取反向互补。
vsearch --sintax result/raw/otus.fa --db ${ea}/usearch/rdp_16s_v16_sp.fa \
--tabbedout result/raw/otus.sintax --sintax_cutoff 0.6
为去除16S rDNA测序中的非特异性扩增和质体污染,我们编写了R脚本otutab_filter_nonBac.R实现选择细菌和古菌(原核生物)、以及去除叶绿体和线粒体并统计比例,输出筛选后并按丰度排序的特征表。输入文件为原始特征表(result/raw/otutab.txt)和物种注释(result/raw/otus.sintax),输出筛选并排序的特征表(result/otutab.txt)、统计污染比例文件(result/raw/otutab_nonBac.txt)和过滤细节(otus.sintax.discard),特征表的格式详见文件或附表2。注:真菌ITS数据,请改用otutab_filter_nonFungi.R脚本,只筛选注释为真菌的序列。查看脚本帮助,请运行Rscript ${ea}/script/otutab_filter_nonBac.R -h。
Rscript ${ea}/script/otutab_filter_nonBac.R \
--input result/raw/otutab.txt \
--taxonomy result/raw/otus.sintax \
--output result/otutab.txt\
--stat result/raw/otutab_nonBac.stat \
--discard result/raw/otus.sintax.discard
5.3筛选特征表对应序的列和物种注释
特征表筛选后,对应的代表序列(otus.fa或附表3)、物种注释信息也需要对应进行筛选。
cut -f 1 result/otutab.txt | tail -n+2 > result/otutab.id
usearch -fastx_getseqs result/raw/otus.fa \
-labels result/otutab.id -fastaout result/otus.fa
awk 'NR==FNR{a[$1]=$0}NR>FNR{print a[$1]}'\
result/raw/otus.sintax result/otutab.id \
> result/otus.sintax
sed -i 's/\t$/\td:Unassigned/' result/otus.sintax
此外,如果上述筛选方案不适合你的研究,如去除的Chloroplast为你研究的对象,可以跳过此步不进行筛选,运行cp result/raw/otu* result/。
接下来对最终的特征表进行统计,结果有助于优化前面的分析方案,以及选择下游分析合适的参数。
usearch -otutab_stats result/otutab.txt \
-output result/otutab.stat
cat result/otutab.stat

图2. USEARCH的特征表统计结果示例
统计结果显示了总读长数量、样本量、特征数量,可以了解特征表的总体数据量和维度信息;接下来是特征表中单元格数量、为0、1和>10的数量和比例,了解特征中频次分布情况;然后是特征在100%、90%和50%样品中发现的数量,展示了特征在样本中的流行频率;最后是样本测序量的分位数,对选择合适的重采样阈值非常有帮助。
我们根据特征表统计结果,将选择合适的参数对特征标进行等量重抽样方式的标准化,以减小由于样本测序量不一致引起的多样性差异,可实现更准确地多样性分析。重采样使用otutab_rare.R脚本调用vegan包(Oksanen等,2007)实现,并计算了6种常用alpha多样性(richness、chao1、ACE、shannon、simpson和invsimpson)指数(vegan.txt或表2)。重采样深度(--depth)参考特征表统计结果(图2)选择,一般默认按最小值重采样。提高采样深度可以保留样本中更大的测序量,但也会剔除低于阈值的样本。因此如果样本测序量波动极大,尽量选择合适的阈值重采样以最大化保留测序量。
mkdir -p result/alpha
Rscript ${ea}/script/otutab_rare.R --input result/otutab.txt \
--depth 32086 --seed 1 \
--normalize result/otutab_rare.txt \
--output result/alpha/vegan.txt
usearch -otutab_stats result/otutab_rare.txt \
-output result/otutab_rare.stat
cat result/otutab_rare.stat
结果显示所有样本重采样后读长数量均为32086。这样特征表可以最大化减少测序量的影响,以便更准确评估多样性。
表2. Alpha多样性指数示例
SampleID | richness | chao1 | ACE | shannon | simpson | invsimpson |
KO1 | 2350 | 2692.008 | 2686.869 | 6.132835 | 0.990308 | 103.1788 |
KO2 | 2316 | 2664.35 | 2661.86 | 6.17406 | 0.991875 | 123.0733 |
KO3 | 1935 | 2278.252 | 2283.343 | 5.828452 | 0.989582 | 95.98662 |
由otutab_rare.R调用vegan包计算的6种常用alpha多样性指数,图中仅展示结果前4行作为示例。
6.Alpha多样性计算
前面在特征表重采样标准化时,计算了6种常用alpha多样性指数。此外,USEARCH的-alpha_div命令可以快速计算18种alpha多样性指数(alpha.txt),各种指数的计算公式和描述详见:http://www.drive5.com/usearch/manual/alpha_metrics.html。这些结果我们常用于结合样品元数据开展组间比较,或箱线图展示组间异同。
usearch -alpha_div result/otutab_rare.txt \
-output result/alpha/alpha.txt
由于测序数据深度对多样性影响较大,有时我们也关注不同测序样量多样性的变化,即可以判断组间差异是否在不同测序深度下稳定存在,同时确定测序量是否饱和并反映出结果较真实的多样性。USEARCH的-alpha_div_rare命令实现快速无放回百分数重采样计算各样本的丰富度(richness/observed_feature,详见alpha_rare.txt或附表4)。结果可进一步可视化为样本稀释曲线,或分组带误差棒的稀释曲线或箱线图。
usearch -alpha_div_rare result/otutab_rare.txt \
-output result/alpha/alpha_rare.txt \
-method without_replacement
Alpha多样性丰富度指数相似只代表物种数量相近,然而其中的物种种类可能完全不同。我们需要制作记录每个组指大于定丰度的物种是否存在的数据格式(表3和附表5),用于组间比较物种共有和特有的情况。可以使用ImageGP在线(http://www.ehbio.com/ImageGP/)选择维恩图(Venn diagram)、集合图(Upset view)或桑基图(Sankey diagram)等方式展示。
表3. 用于比较各组特征共有/特有的数据示例
特征ID | 分组ID |
ASV_1 | All |
ASV_1 | KO |
ASV_1 | OE |
ASV_1 | WT |
ASV_2 | KO |
… | … |
我们通常结合元数据计算各组的丰度均值,如以Group列为分组信息计算原始计数的相对丰度并求组均值。
Rscript ${ea}/script/otu_mean.R --input result/otutab.txt \
--design metadata.txt \
--group Group --thre 0 \
--output result/otutab_mean.txt
因为特征的数量较大,而且低丰度的特征是否存在偶然性较大,准确性不高且与测序噪音无法区分。因此筛选大于某一丰度阈值结果,可实现数据降维并保留数据的主体,然后用于组间比较共有和特有的情况。如以平均丰度> 0.1%为阈值,可选0.5%或0.05%,得到每个组中符合条件的特征(表3)。
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {for(i=2;i<=NF;i++) a[i]=$i;} \
else {for(i=2;i<=nf;i++) i="">0.1) print $1, a[i];}}' \
result/otutab_mean.txt > result/alpha/otu_group_exist.txt
7.Beta多样性计算
Beta多样性是群落整体结构的降维分析方法,需要基于特征表计算样本间的各种距离矩阵。常用的Unifrac算法(Lozupone等,2010)考虑物种间的进化距离,这里我们使用usearch的-cluster_agg命令基于代表序列获得进化树,然后再使用-beta_div基于特征表和进化树计算多种矩阵矩阵,包括bray_curtis, euclidean, jaccard, manhatten, unifrac等,每类矩阵还分为有或无(binary)权重两种(附表6展示Bray-Curtis距离矩阵示例)。
mkdir -p result/beta/
usearch -cluster_agg result/otus.fa -treeout result/otus.tree
usearch -beta_div result/otutab_rare.txt -tree result/otus.tree \
-filename_prefix result/beta/
8.物种注释分类汇总
前在特征表筛选时已经对特征序列完成了物种注释,并根据注释进行筛选。物种注释存在命名混乱、分类级不完整和名称缺失等问题。我们先对格式进行调整方便开展分析。调整物种注释为特征ID和7级分类注释的两列格式(表4和附表7)。注意7级分类可能存在不完整的情况,可能是该特征没有相近种的报导,也可能是参考物种注释自身不完善。
cut -f 1,4 result/otus.sintax \
|sed 's/\td/\tk/;s/:/__/g;s/,/;/g;s/"//g;s/\/Chloroplast//' \
> result/taxonomy2.txt
表4. 物种注释2列格式示例
特征ID | 分组ID |
ASV_1 | k__Bacteria;p__Actinobacteria;c__Actinobacteria;o__Actinomycetales;f__Thermomonosporaceae |
ASV_2 | k__Bacteria;p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Comamonadaceae;g__Pelomonas;s__Pelomonas_puraquae |
ASV_3 | k__Bacteria;p__Proteobacteria;c__Betaproteobacteria;o__Burkholderiales;f__Comamonadaceae;g__Pelomonas;s__Pelomonas_puraquae |
物种注释的7级分类,也提供了特征表多角度降维分析的可能,可以把特征表按物种注释信息合并为门、纲、目、科、属级别的表,以进行更容易与现在科学发现结合的讨论。首先将物种注释转化为7级分类的8列表格(表5和附表8),其中缺失的分类级别填充为未分类(Unassigned)。
awk 'BEGIN{OFS=FS="\t"}{delete a; a["k"]="Unassigned";a["p"]="Unassigned";a["c"]="Unassigned";a["o"]="Unassigned";a["f"]="Unassigned";a["g"]="Unassigned";a["s"]="Unassigned";\
split($2,x,";");for(i in x){split(x[i],b,"__");a[b[1]]=b[2];} \
print $1,a["k"],a["p"],a["c"],a["o"],a["f"],a["g"],a["s"];}' \
result/taxonomy2.txt > temp/otus.tax
sed 's/;/\t/g;s/.__//g;' temp/otus.tax|cut -f 1-8 | \
sed '1 s/^/OTUID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' \
> result/taxonomy.txt
表5. 物种注释8列格式示例
OTUID | Kingdom | Phylum | Class | Order | Family | Genus | Species |
ASV_1 | Bacteria | Actinobacteria | Actinobacteria | Actinomycetales | Thermomonosporaceae | Unassigned | Unassigned |
ASV_2 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | Pelomonas | Pelomonas_puraquae |
ASV_3 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Rhizobacter | Rhizobacter_bergeniae |
接下来对特征表按各级别进行分类汇总,获得门、纲、目、科和属水平上的特征表(附表9门水平分类汇总表),即可以直接用来绘制物种组成图,也可以进一步从更多角度分析组间差异或挖掘生物标志物。
for i in p c o f g;do
usearch -sintax_summary result/otus.sintax \
-otutabin result/otutab_rare.txt -rank ${i} \
-output result/tax/sum_${i}.txt
done
sed -i 's/(//g;s/)//g;s/\"//g;s/\#//g;s/\/Chloroplast//g' result/tax/sum_*.txt
9.有参分析和功能预测
一些功能注释数据库,如PICRUSt (Langille等,2013)、BugBase (Ward等,2017)等的输入文件必须是基于GreenGenes数据库生成的特征表。此处采用vsearch的--usearch_global命令比对扩增子数据至GreenGenes数据库即可获得有参分析的特征表(gg/otutab.txt),该结果可作为主流功能预测软件的输入文件开展分析。
vsearch --usearch_global temp/filtered.fa --db ${ea}/gg/97_otus.fasta \
--otutabout result/gg/otutab.txt --id 0.97 --threads 12
注意:如果使用PICRUSt2可直接使用第4步中的序列(otus.fa)和第5步中的特征表(result/otutab.txt)作为输入文件开展分析。
10.空间清理及数据提交
整个分析过程会占用原始数据大小数10倍的空间。项目结果分析结束可以删除整个temp文件夹。分析取得阶段性成果,也要及时清楚不常用的中间文件节省空间,如fastq文件。这个习惯对于团队共享存储时尤其重要,否则硬盘空间耗尽,所有任务都会立即停止。
rm -rf temp/*.fq
短期不用数据库压缩节省空间。
gzip ${ea}/usearch/rdp_16s_v16_sp.fa
gzip ${ea}/gg/97_otus.fasta
原始序列统计md5值,用于数据提交(附表10)。
cd seq
md5sum *_1.fq.gz > md5sum1.txt
md5sum *_2.fq.gz > md5sum2.txt
paste md5sum1.txt md5sum2.txt | awk '{print $2"\t"$1"\t"$4"\t"$3}' | \
sed 's/*//g' > ../result/md5sum.txt
rm md5sum*
cd ..
cat result/md5sum.txt
11.STAMP和LEfSe软件输入文件准备
STAMP是常用的图型界面特征差异比较软件(Parks等,2014),操作简单,同时支持主流操作系统(Windows/Linux/Mac,在Windows中安装最方便),软件可从其主页https://beikolab.cs.dal.ca/software/STAMP 获取。我们提供了format2stamp.R脚本,可以基于特征表、物种注释信息快速获得界、门、纲、目、科、属、种、OTU/ASV的STAPM输入格式兼容的特征表,同时提供按丰度过滤的参数。如按万分之一相对丰度过滤生成STAMP输入文件的代码如下:
Rscript ${ea}/script/format2stamp.R -h
mkdir -p result/stamp
Rscript ${ea}/script/format2stamp.R --input result/otutab.txt \
--taxonomy result/taxonomy.txt --threshold 0.01 \
--output result/stamp/tax
LEfSe是常用的生物标记鉴定软件(Segata 等,2011),支持多组比较。其输入文件格式是整合了样本分组信息、界、门、纲、目、科、属层面相对丰度的结果。同时为了展示可读性的进化分枝树图形,还需要对特征表进行筛选。我们提供了format2lefse.R脚本,可以一步生成LEfSe要求的输入文件,同时提供按丰度过滤的参数。如按千分之一(threshold)丰度筛选以控制作图中的进化分枝数量有较好的可读性,代码如下:
mkdir -p result/lefse
Rscript ${ea}/script/format2lefse.R --input result/otutab.txt \
--taxonomy result/taxonomy.txt --design result/metadata.txt \
--group Group --threshold 0.1 \
--output result/lefse/LEfSe
结果文件可以在软件官网(http://huttenhower.sph.harvard.edu/galaxy)或我们建立的国内备份站ImageGP(http://www.ehbio.com/ImageGP)开展在线分析。结果的进化分枝图中分枝过密和/或文字重叠严重,可进一步提高丰度阈值以减少分枝数量,反之同理。
常见问题
1.软件、数据库下载慢或无法下载
由于国际带宽和站点的速度限制等原因,很多国外数据库下载缓慢甚至无法下载。宏基因组公众号团队建立了微生物组领域的扩增子和宏基因组常用软件和数据库的国内备份站点,方便同行下载和使用。站点1. 国家微生物科学数据中心的数据下载页面—工具资源下载栏目(http://nmdc.cn/datadownload)即为宏基因组团队与中科院微生物所共同维护的站点之一,提供宏基因组常用软件、数据库的FTP下载链接。站点2. 由刘永鑫的GitHub中《微生物组数据分析与可视化实战》专著的大数据下载页面(https://github.com/YongxinLiu/MicrobiomeStatPlot/blob/master/Data/BigDataDownlaodList.md) 提供有常用资源下载百度云链接和HTTP下载链接。
R包的批量安装,需要在RStudio中查看R包所在位置,然后替换下载的R包合辑,详细操作见—个人电脑搭建微生物组分析平台(Win/Mac)。
2.ITS物种注释数据库UNITE使用时报错
UNITE数据库官方提供的数据格式有时存在错误。主要是分类级空缺的问题,我们使用sed命令对utax8.2数据库进行调整,示例如下。
sed -i 's/,;/,Unnamed;/;s/:,/:Unnamed,/g' utax_reference_dataset_all_04.02.2020.fasta
3.文件Phred质量错误—Fastq质量值64转33
Illumina测序的Fastq格式文件中序列的质量值通常为Phred33格式,典型特点为以大写字母为主。有时测序服务提供商也会提供旧版Phred64格式的Fastq文件,直接使用会提示Phred编码错误,我们需要使用vsearch的--fastq_convert转换Phred64为常用的Phred33格式。此外可选fastp(Chen等, 2018)实现格式转换。
vsearch --fastq_convert test_64.fq \
--fastqout test.fq \
--fastq_ascii 64 --fastq_asciiout 33
4.单端序列改名
如果是单端测序数据或已经合并后的单端FASTQ序列样本文件,需要按样本名重命名每条序列,才能进行下游分析,否则将无法区分序列的样本来源。我们使用vsearch --fastq_convert命令中的--relabel参数对序列按样本重命名,以WT1样本为例。
vsearch --fastq_convert test.fq --fastqout WT1.fq --relabel WT1.
5.Fasta序列取反向互补
物种注释时发现序列全为反向(-),表明序列的方向有错误,可用vsearch的--fastx_revcomp命令调整。
vsearch --fastx_revcomp filtered_test.fa --fastaout filtered.fa
致谢
本项目由中国科学院前沿科学重点研究项目(编号:QYZDB-SSW-SMC021)、国家自然科学基金面上项目(编号:31772400) [Supported by the Key Research Program of Frontier Sciences of the Chinese Academy of Science (No. QYZDB-SSW-SMC021), the National Natural Science Foundation of China (No. 31772400)]支持。此分析流程在我之前的综述中被提及(刘永鑫等,2019; Liu等,2020),本文是发表的详细使用方法和常见问题解决的经验。
参考文献
1.刘永鑫, 秦媛, 郭晓璇, 白洋 (2019). 微生物组数据分析方法与应用. 遗传 41(9): 845-826.
2.Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Zech Xu, Z., Kightley, E. P., Thompson, L. R., Hyde, E. R., Gonzalez, A. and Knight, R. (2017). Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2(2): e00191-00116.
3.Chen, Q., Jiang, T., Liu, Y. X., Liu, H., Zhao, T., Liu, Z., Gan, X., Hallab, A., Wang, X., He, J., Ma, Y., Zhang, F., Jin, T., Schranz, M. E., Wang, Y., Bai, Y. and Wang, G. (2019). Recently duplicated sesterterpene (C25) gene clusters in Arabidopsis thaliana modulate root microbiota. Sci China Life Sci 62(7): 947-958.
4.Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i884-i890.
5.Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., Brown, C. T., Porras-Alfaro, A., Kuske, C. R. and Tiedje, J. M. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42(Database issue): D633-642.
6.Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19): 2460-2461.
7.Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10(10): 996-998.
8.Edgar, R. C. and Flyvbjerg, H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31(21): 3476-3482.
9.Huang, A. C., Jiang, T., Liu, Y. X., Bai, Y. C., Reed, J., Qu, B., Goossens, A., Nutzmann, H. W., Bai, Y. and Osbourn, A. (2019). A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364(6440): eaau6389.
10.Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., Clemente, J. C., Burkepile, D. E., Vega Thurber, R. L., Knight, R., Beiko, R. G. and Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9): 814-821.
11.Liu, Y. X., Qin, Y., Chen, T., Lu, M., Qian, X., Guo, X. and Bai, Y. (2020). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 11.
12.Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. and Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J 5(2): 169-172.
13.McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., Andersen, G. L., Knight, R. and Hugenholtz, P. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6(3): 610-618.
14.Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., Kennedy, P., Picard, K., Glockner, F. O., Tedersoo, L., Saar, I., Koljalg, U. and Abarenkov, K. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47(D1): D259-D264.
15.Oksanen, J., Kindt, R., Legendre, P., O’Hara, B., Stevens, M. H. H., Oksanen, M. J. and Suggests, M. (2007). The vegan package. Commun Ecol Pack 10: 631-637.
16.Qian, X. B., Chen, T., Xu, Y. P., Chen, L., Sun, F. X., Lu, M. P. and Liu, Y. X. (2020a). A guide to human microbiome research: study design, sample collection, and bioinformatics analysis. Chin Med J (Engl) 133(15): 1844-1855.
17.Qian, X., Liu, Y. X., Ye, X., Zheng, W., Lv, S., Mo, M., Lin, J., Wang, W., Wang, W., Zhang, X. and Lu, M. (2020b). Gut microbiota in children with juvenile idiopathic arthritis: characteristics, biomarker identification, and usefulness in clinical prediction. BMC Genomics 21(1): 286.
18.Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J. and Glockner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue): D590-596.
19.Rognes, T., Flouri, T., Nichols, B., Quince, C. and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4: e2584.
20.Wang, Y., Song, F., Zhu, J., Zhang, S., Yang, Y., Chen, T., Tang, B., Dong, L., Ding, N., Zhang, Q., Bai, Z., Dong, X., Chen, H., Sun, M., Zhai, S., Sun, Y., Yu, L., Lan, L., Xiao, J., Fang, X., Lei, H., Zhang, Z. and Zhao, W. (2017). GSA: Genome Sequence Archive*. Genomics Proteomics Bioinformatics 15(1): 14-18.
21.Ward, T., Larson, J., Meulemans, J., Hillmann, B., Lynch, J., Sidiropoulos, D., Spear, J. R., Caporaso, G., Blekhman, R., Knight, R., Fink, R. and Knights, D. (2017). BugBase predicts organism-level microbiome phenotypes. bioRxiv: 133462.
22.Zhang, J., Liu, Y.-X., Zhang, N., Hu, B., Jin, T., Xu, H., Qin, Y., Yan, P., Zhang, X., Guo, X., Hui, J., Cao, S., Wang, X., Wang, C., Wang, H., Qu, B., Fan, G., Yuan, L., Garrido-Oter, R., Chu, C. and Bai, Y. (2019). NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol 37(6): 676-684.
23.Zhang, J., Zhang, N., Liu, Y. X., Zhang, X., Hu, B., Qin, Y., Xu, H., Wang, H., Guo, X., Qian, J., Wang, W., Zhang, P., Jin, T., Chu, C. and Bai, Y. (2018). Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci China Life Sci 61(6): 613-621.
24.Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30(21): 3123-3124.
25.Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S. & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology 12(6): R60.
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 2462
2462

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









