






点击蓝字 关注我们
iNAP2.0:代谢互补在微生物网络分析中的应用

方法论文
● 期刊:iMeta(IF 23.7)
● 原文链接DOI: https://doi.org/10.1002/imt2.235
● 2024年9月2日,中国科学院生态环境研究中心邓晔组在iMeta在线发表了题为“iNAP 2.0: Harnessing metabolic complementarity in microbial network analysis”的文章。
● 本研究中提出了更新的iNAP2.0,其最重要的进步是整合了代谢模型的构建与分析模块,用于根据宏基因组测序数据进行微生物代谢互作的研究。
● 第一作者:彭玺
● 通讯作者:冯凯(kaifeng@rcees.ac.cn)、邓晔(yedeng@rcees.ac.cn)
● 合作作者:杨兴盛、何晴、赵博、李曈、王尚
● 主要单位:中国科学院生态环境研究中心、中国科学院大学资源与环境学院
亮 点
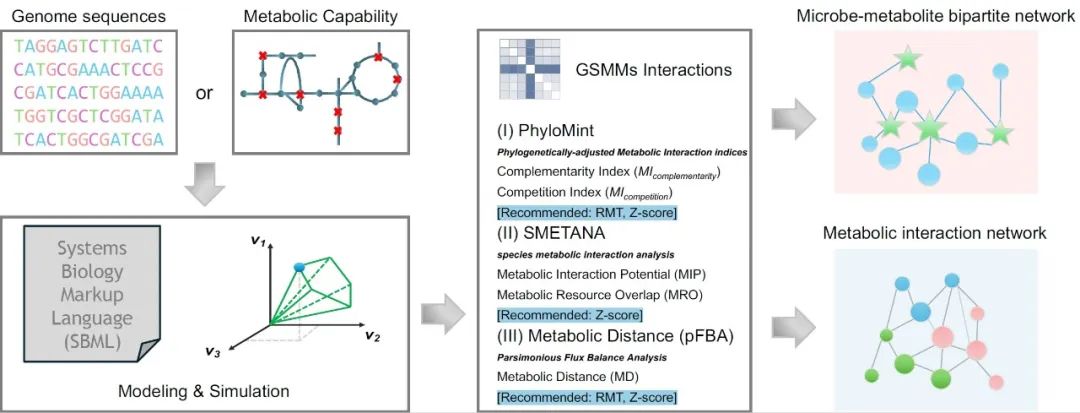
● 更新后的iNAP 2.0可以计算各种代谢互补与竞争系数,如PhyloMint指数、SMETANA(物种代谢相互作用分析)分数与基于pFBA的代谢距离,并提供了代谢互作网络的构建与分析模块;
● iNAP 2.0创新性地使用了随机矩阵理论模型(RMT)来选择筛选代谢互作网络的阈值;
● iNAP 2.0中的PhyloMint PTM程序可以显示微生物相互作用间的可能转移的代谢物,并将它们和微生物网络一起构建微生物-代谢物二分网络。
摘 要
随着宏基因组测序技术的广泛使用,研究微生物生态网络的新视角应运而生,基于相关性的共现网络所无法预测的代谢证据得到了补充。本研究中提出了更新的iNAP2.0,其最重要的进步是整合了代谢模型的构建与分析模块,用于根据宏基因组测序数据进行微生物代谢互作的研究。iNAP2.0建立的代谢互作分析流程包括四个模块:(I)准备基因组规模代谢模型;(II)推断基因组规模代谢模型的成对相互作用;(III)构建代谢相互作用网络;(IV)分析代谢相互作用网络。无论是从宏基因组组装基因组(MAG)开始,或是从完整基因组开始,iNAP2.0提供了多种方法来量化模型之间的代谢互补性的潜力与趋势,包括基于系统发育距离调整的PhyloMint工具、物种代谢相互作用分析方法SMETANA以及基于简约流平衡分析(pFBA)的代谢距离计算等。另外,iNAP2.0集成了随机矩阵理论模型(RMT)方法来客观地筛选代谢互作网络的合适阈值。最后,iNAP2.0的一个重要工具是识别物种之间潜在可转移的代谢物,并将它们呈现为链接代谢互补网络中微生物节点的中间节点。iNAP2.0已在https://inap.denglab.org.cn上开放注册,并提供了测试数据供研究者们使用。
视频解读
Bilibili:https://www.bilibili.com/video/BV1svxDeGEWj/
Youtube:https://youtu.be/uZj-tXJNISU
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
微生物生态网络或微生物互作网络为微生物生态学研究提供了有价值的见解,然而,它们依赖于统计推断和无法推断功能细节的非常有限。然而,随着宏基因组数据分析的迅速发展,基于功能性状或代谢分析的方法可以更深入地了解微生物生态网络,同时,生物信息学家们开发了能预测或模拟微生物间代谢交换或代谢互补的各种建模方法。这些方法均依赖于微生物基因组(例如参考基因组、宏基因组组装基因组或单扩增基因组)及其基因组规模代谢模型中 编码的信息。不同的方法对于这些信息的使用程度不尽相同,一些方法仅关注反应和代谢物的类型,而另一些方法则考虑微生物的代谢效率或生长速率等因素。具体来说,PhyloMint关注成对基因组中代谢物的类型,SMETANA关注群落中代谢资源的重叠与交换(可以是两个及以上的物种,包含更高阶的相互作用),代谢距离的计算则涉及模拟生长或能量的代谢模型中的代谢通路。尽管这些建模工具能够构建并分析基因组规模代谢模型,但他们通常依赖专门的软件或编程语言,例如Python、MATLAB与命令行界面,这为非生物信息的专业工作者造成了困难。同时模型间的代谢相互作用缺少一个方法筛选构建网络的数值。为解决这些问题,我们升级了iNAP至iNAP 2.0,其中包括了自动化构建基因组规模代谢模型的工具CarveMe、计算代谢互作系数的PhyloMint、SMETANA、Cobrapy等工具。以此,iNAP 2.0可以作为基于相关性的微生物共现网络的重要补充方法。
工作流程的概览与实现
iNAP2.0新增的代谢建模流程允许用户从基因组序列开始分析(图1)。该流程利用Prokka预测编码序列,并使用CarveMe自动化构建基因组规模代谢模型(GSMMs)。或者,用户也可以使用ModelSEED等工具手动构建GSMMs,或者直接从如VMH(Virtual Metabolic Human)数据库导入提前构建好的模型。对于微生物间代谢相互作用的分析,iNAP 2.0提供了三种方法:PhyloMint(竞争/互补性指数)、SMETANA分数和基于简约流平衡分析(pFBA)的代谢距离。值得注意的是,PhyloMint指数突出了在计算过程中考虑的潜在可转移代谢物。iNAP 2.0提供了RMT方法用于找到统计显著性的客观决定阈值,以构建和分析所得代谢相互作用网络的拓扑属性。与iNAP相同,iNAP2.0继续使用Galaxy框架,并提供了详细的操作说明,概述了分析步骤。本操作说明解释了每种建模方法的参数选择和结果,以易于研究者们按照顺序使用工具并进行分析。
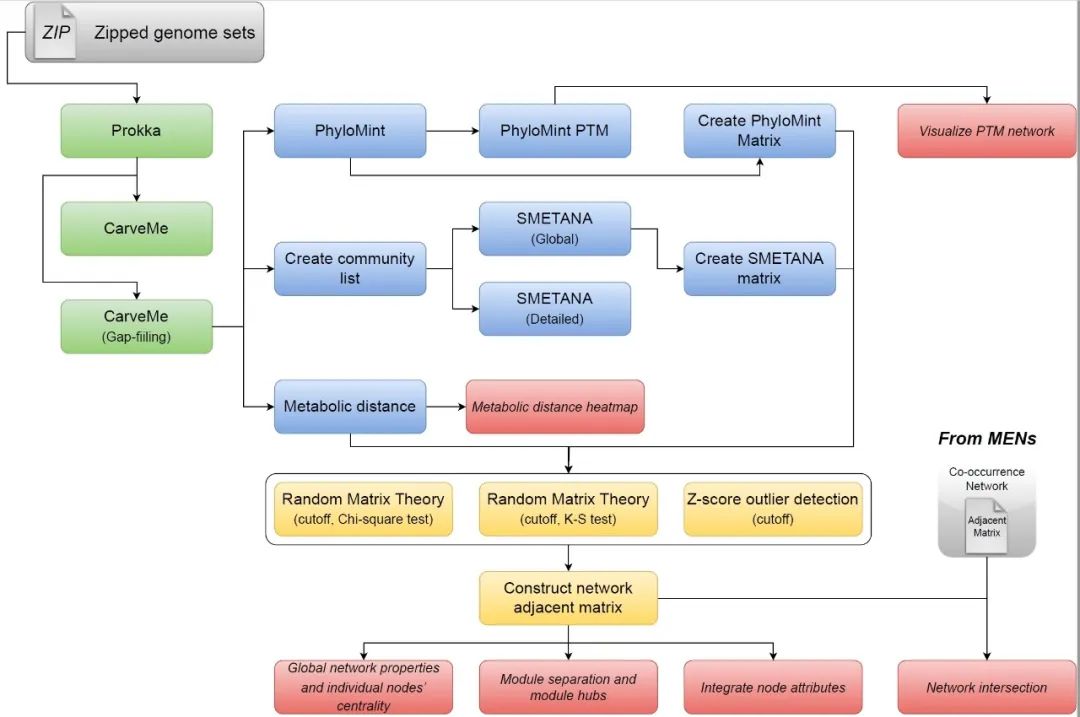
图1. 代谢模型分析和代谢互作网络构建的工作流程
PTM:潜在可转移代谢物;K-S test:Kolmogorov–Smirnov test;MEN:分子生态网络。
第一部分:准备基因组规模代谢模型
根据输入文件格式的要求,用户需要上传zip压缩的基因组集或蛋白序列集。iNAP2.0提供了两组Demo基因组集存储在User Panel—Shared Data—Data Libraries—Metabolic Modeling Demo Datasets。
Box 1:输入文件格式要求
● 压缩的基因组集(.zip):该文件应包含所有要分析的基因组序列文件(.fasta/.fa)。确保所有序列文件被直接压缩,而不是存储在文件夹中再压缩。每个序列文件名应唯一,且不以数字开头,不含空格、连字符或其他特殊字符(可以使用下划线)。如果计划使用SMETANA分析,则上传的基因组文件数量不应超过300个,因为该方法的计算资源消耗巨大;
● 预测的蛋白质序列(.zip):该文件应包含基因组序列对应的蛋白质序列文件(.faa)。可以使用Prokka工具获得此文件,或从预先获得或参考数据库中下载。压缩与命名要求与上述相同;
● 用于Gap-filling的培养基文件(.txt/.tabular)。在使用步骤2-B时,除了默认的5种培养基外,用户可以上传自定义的培养基。培养基描述文件中的化合物名称必须与BiGG数据库一致。
步骤1 Prokka:iNAP 2.0使用Prokka(默认参数)进行基因组注释。或者,用户也可以使用如Prodigal或EGGNOG-mapper之类的工具来代替此步骤。输出为压缩的蛋白质序列文件。
步骤2-A CarveMe:iNAP 2.0使用CarveMe来快速且自动化的构建基因组规模代谢模型(GSMMs)。这一步使用步骤1的输出——压缩的蛋白质序列文件,或者预先下载的压缩的注释蛋白质集作为输入。输出格式为sbml-fbc2,它可以与大多数基于约束的建模工具兼容。在CarveMe构建的GSMM中,反应通量的上下限直接调用Cobra中的默认值(cobra_default_ub, cobra_default_lb),分别设置为为1000和-1000 mmol/gDW/h。这一步的输出文件是压缩的代谢模型文件(.xml)。
步骤2-B CarveMe (gap filling):当使用CarveMe构建GSMM时,程序会计算各种反应的得分。它使用混合整数线性规划(MILP)以决定哪些反应应该包含在最终模型中。因此,由于分箱或注释的限制,自然环境中的微生物模型可能缺乏某些反应。我们建议使用gap filling来修正从环境宏基因组组装基因组(MAGs)中得到的模型。CarveMe为gap filling提供了五种预设的培养基配方(默认值为LB培养基),允许模型在相应的培养基上进行生长模拟。用户也可以自定义设置培养基配方,例如肠道微生物组模型的diet组成。需要注意的是,由于培养大多数微生物的困难以及营养丰富的培养基的局限性,为环境微生物组定义培养基实际上并不容易。使用营养丰富的培养基进行gap filling可以缓解过度纠正(over gap-filling)的问题。我们也推荐另外使用一些工具,例如基于深度学习方法的CHESHIRE(CHEbyshev Spectral HyperlInk pREdictor)来作为gap-filling的辅助工具,从而优化代谢模型。输入和输出文件与步骤2-A相同。建议用户在说明结果中声明用于gap filling的培养基组成,以保证模型的可复现性。
Box 2:关于GSMM质量的说明
虽然GSMM是一个强大的工具,但它并非没有局限性,有几个因素会影响模型构建的质量。尽管我们没有提供评估GSMM的质量的方法,GSMM的可信度可以通过以下策略来改善。
1)GSMM的可靠性与所使用的基因组质量密切相关。基因组的质量各不相同,如完整的参考基因组和宏基因组组装基因组(MAGs)。在条件允许的情况下,应优先使用完整的参考基因组进行GSMM重建。然而,鉴于许多微生物无法在实验室条件下分离和培养,MAGs可以作为大多数情况下,尤其是在宏基因组数据时。尽管如此,必须强调MAGs的质量,特别是其完整性,会显著影响模型的准确性。因此,在利用MAGs用于GSMM构建时应采用更严格的标准;
2)GSMM构建工具的选择也会影响模型的质量。Mendoza等人对GSMM工具进行了系统评估,为用户提供了四个关键标准:可发现性(findability)、可达性(accessible)、交叉操作性(interoperability)和复用性(reusability)。基于这些标准,像CarveMe这样的工具构建的模型与手动建立的模型在反应集上表现出高度相似,因此得到选用。在本操作说明中,我们鼓励用户探索各种模型构建工具。然而,研究人员也应该对可能出现的假结果保持警惕,尤其是对于这类基因水平的研究。
第二部分:推断GSMMs间的成对相互作用
iNAP 2.0提供了三种方法来量化GSMMs之间的代谢相互作用:PhyloMint、SMETANA和基于pFBA的代谢距离。与基于物种丰度的共现网络类似,这些代谢相互作用指数可以通过筛选并构建成微生物代谢网络。本节将解释每种方法的原理和解释。
方法1:PhyloMint
PhyloMint使用GSMMs来预测基因组之间的竞争和互补性(MIcompetition和MIcomplementarity)的代谢指数。对于每个GSMM,首先预测一组种子化合物,即一组无法在模型中内源合成的最小化合物子集(代谢模型中的强连通组分,SCC)。然后,使用公式计算基因组对之间的竞争和互补性指数。
步骤3 PhyloMint:程序计算n个输入GSMMs(步骤2-A/B的输出)之间的MIcompetition/MIcomplementarity,生成n2组指数。这些指数显示了两个模型在代谢功能上共享部分以及一个模型能利用另一个模型释放的物质的程度。参数MaxCC表示SCC集合的最大大小(默认值:5,仅影响竞争系数)。PhyloMint的结果是一个包含4列的表格,显示基因组对及其MIcompetition和MIcomplementarity。
步骤4 PhyloMint PTM:按照计算公式定义,MIcomplementarity指数代表一个模型能利用另一个模型的代谢物质的潜力。潜在可转移代谢物(PTMs)定义为一个模型的种子化合集和另一个模型的非种子化合集的交集。这一步以压缩模型集(步骤2-A/B的输出)和PhyloMint结果(步骤3的输出)作为输入,并以表格文件的形式输出这些物质,显示每个化合物的供体和受体以及它们在BiGG数据库中的命名和完整索引。必须注意,如果使用的GSMMs不是由CarveMe生成的,文件扩展名可能是SBML格式,用户需要在这一步更改格式或扩展名。
步骤5 Create PhyloMint Matrix:这一步将步骤4生成的结果从表格形式转换为矩阵形式,用于网络阈值确定。特别是,我们为不对称的MI指数提供两种处理模式:(1)保留原始值,即直接将步骤4的结果转换为不对称矩阵。使用此矩阵构建的代谢相互作用网络将是有向的;(2)选择每对模型的成对指数中的较大值来表示模型之间的相互作用强度,以使得生成的矩阵是对称的,使用它构建的代谢相互作用网络将是无向的。我们将(2)设为默认,并推荐使用基于RMT的方法进行网络阈值确定。
方法2:SMETANA
SMETANA是一个通过计算群落中代谢资源的重叠和最小生长需求代谢物来量化群落内的竞争和互补性的工具。SMETANA可以模拟由两个及以上GSMMs组成的群落,因此程序(步骤7和8)需要一个表格来指示给定模型集中哪些GSMMs属于一个群落。在iNAP 2.0平台上,SMETANA分析的模型限制为300个。
步骤6 Create Community List:为了计算GSMMs之间的成对相互作用,iNAP 2.0提供了一个给定模型集中所有GSMMs成对的所需表格(步骤2-A/B的输出),这将用作步骤7(-A/B)和8(-A/B)的输入。
步骤7-A SMETANA Global:对于给定的群落,SMETANA定义了两个指数,代谢资源重叠(MRO)和代谢互作潜力(MIP),分别代表群落的竞争和互补性水平。这些指数的具体定义和计算已经在以往文献中详细描述。可以使用SMETANA的global模式计算这些指数。这一步的输入应是压缩的基因组集和在步骤6输出的表格。
步骤7-B Iterative SMETANA Global:SMETANA的开发者已经确认它的一个bug,即每次运行的结果可能会略有不同。这种不一致性是由于CPLEX solver的solution pool特性造成的。开发者建议可以多次运行并计算均值以代表最终结果。为了实现这一点,我们设计了此步骤,将步骤7-A运行2-10次,并输出平均结果。
步骤8-A SMETANA Detailed:除了使用MRO/MIP量化群落的代谢互作外,SMETANA还提供了detailed模式来计算一系列指数,以进一步量化物种间的相互作用。这些指数包括:物种偶联得分(SCS)、代谢物吸收得分(MUS)、代谢物产生得分(MPS)以及结合了三个指数的SMETANA得分,以代表群落中物种间依赖性的总和。本步骤的输入要求与步骤7-A一致。
步骤8-B Iterative SMETANA Detailed:重复计算的原因与步骤7-B相同。
步骤9 Create SMETANA Matrix:这一步将SMETANA MIP/MRO的结果从表格转换为两个矩阵,用于网络阈值确定。该步骤需要群落列表(步骤6的输出)和SMETANA global模式的结果(步骤7-A/B的输出)作为输入文件。
方法3:代谢距离(Metabolic Distance)
先前的研究表明,代谢距离(代谢不相似性)在形成和决定微生物群落中的协同作用中至关重要。
步骤10 Metabolic distance:此步骤根据Giri等人描述的方法计算GSMMs之间的成对代谢距离。这一步的输入是压缩的模型集(步骤2-A/B的输出)。具体来说,该程序首先对每个模型进行流平衡分析(FBA),以反应为优化(最大化)biomass反应通量(目标函数,通常用于代表生长速率反应)。该步骤也可以选择优化其他目标函数,如ATP产量,代表不同的代谢策略。默认设置下,iNAP 2.0选择CarveMe中的“Growth”反应,代表由CarveMe生成的GSMMs中的增长率反应。随后,优化的生物量反应通量被固定,进行简约FBA(pFBA)以最小化每个模型中绝对通量之和,同时约束目标函数(如生物量生产)达到初始FBA获得的最优值。随后,选择在至少一个模型中通量不为零的反应作为代表,生成模型的通量向量,并计算它们之间的欧几里得距离来表征代谢距离。这一步骤有3个输出:2个距离矩阵(原始欧几里得距离和标准化欧几里得距离)和原始欧几里得距离的3列表格(模型A/模型B/欧几里得距离)。最先考虑使用原始欧几里得距离。在矩阵包含许多相差数量级的数值(当同时输入用不同通量上下限的设置构建的模型时会发生,我们非常不建议用这样的模型集)的情况下,可以考虑使用标准化欧几里得距离矩阵。
第三部分:构建代谢相互作用网络
不同的代谢相互作用指数具有不同的数值形式和值域。iNAP 2.0提供了多种方法来确定网络阈值,包括基于随机矩阵理论(RMT)的方法。
步骤11 RMT (cutoff, Chi‐square test):此步骤可以使用PhyloMint(步骤5)和代谢距离(步骤10)生成的邻接矩阵作为输入。由于SMETANA指数的值不符合RMT方法的要求,建议使用Z-score方法(见步骤13)。目前,这一步只接受对称矩阵作为输入,且矩阵中的值应标准化到0和1之间。
步骤12 RMT (cutoff, Kolmogorov-Smirnov test):此步骤和步骤11均使用基于RMT的方法来确定网络阈值。区别在于,第11步使用了卡方检验,而这一步使用了Kolmogorov–Smirnov检验。与卡方检验相比,Kolmogorov–Smirnov检验预期会给出更宽松的阈值,这在处理类似于MIcompetition和MIcomplementarity这样的代谢指数时会更加实用。
步骤13 Z-score outlier detection:iNAP 2.0提供了一种基于Z-score异常值检测方法,用于过滤邻接矩阵中的相互作用,以构建网络。这种方法已用于基于PhyloMint MIcompetition和MIcomplementarity指数构建网络。然而,我们建议将这种方法用于SMETANA指数(例如,MIP)和代谢距离(标准化之前),因为它们可能不符合RMT方法的要求。这一步的输入是PhyloMint(步骤5)、SMETANA全局(步骤7-A/B)或代谢距离(步骤10)生成的邻接矩阵。我们提供了标准的和modified的Z-score公式用于异常值检验:
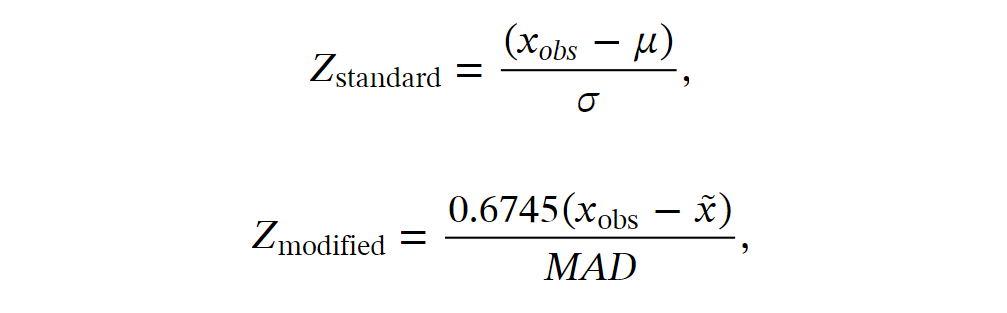

步骤14 Construct Network Adjacent Matrix:在选择了适当的方法来确定网络过滤的阈值(由步骤11、12或13选择)之后,将原始邻接矩阵和选定的阈值输入这一步,它将生成代表最终代谢相互作用网络的邻接矩阵。
第四部分:分析代谢相互作用网络
所有已介绍的iNAP原有的工具均可用于分析代谢相互作用网络,例如网络全局属性、节点中心性、模块化与模块核心节点的识别等。此外,iNAP2.0也新增了一些代谢相互作用网络的新分析工具。
步骤15 Global network properties and individual nodes’ centrality:此步骤使用步骤14得到的代谢相互作用邻接矩阵作为输入。生成两个结果表格文件:(1)网络全局属性文件,包括许多参数,如平均节点度数、平均聚类系数和网络密度,这些参数充分描述了代谢相互作用网络的属性;(2)节点属性文件,包括所有节点的属性,如节点度、介数等。
步骤16 Module separation and module hubs:这一步同样需要使用步骤14得到的代谢相互作用邻接矩阵作为输入。它计算网络的模块性和将节点分配到不同的模块。基于模块化的结果,将计算每个节点的模块内连通性(zi)和模块间连通性(Pi)。基于zi - Pi分布,可以识别核心节点,如模块核心、连接核心和网络核心节点。
步骤17 Integrate node attributes:节点中心性表格(步骤15的输出)、节点的模块连通性表格(zi-Pi值和网络中的角色,步骤16的输出)以及节点的分类注释表格(可选项,由用户上传,制表符分隔的.txt文件,第一列为节点ID,参见iNAP 2.0中的示例文件)是微生物网络注释信息的重要组成部分。这一步可以将上述三个文件合并,用于后续分析或作为可视化的注释文件。
步骤18 Network intersection:此步骤将比较两个网络的邻接矩阵,并识别它们的共同边所组成的子网络,并输出子网络的邻接矩阵与边列表。
步骤19 Visualize the PTM network:这一步使用步骤5(PhyloMint PTM)的结果作为输入,将特定网络中的潜在可转移代谢物转化为有向的微生物-代谢物二分网络(如图2A)。输入中存储的所有代谢物注释将被整合并输出在节点属性表中。
步骤20 Metabolic distance heatmap:这一步使用步骤10(Metabolic distance)得到的代谢距离矩阵生成热图,这有助于显著识别微生物群落中具有独特的代谢特征的物种(如图2B)。
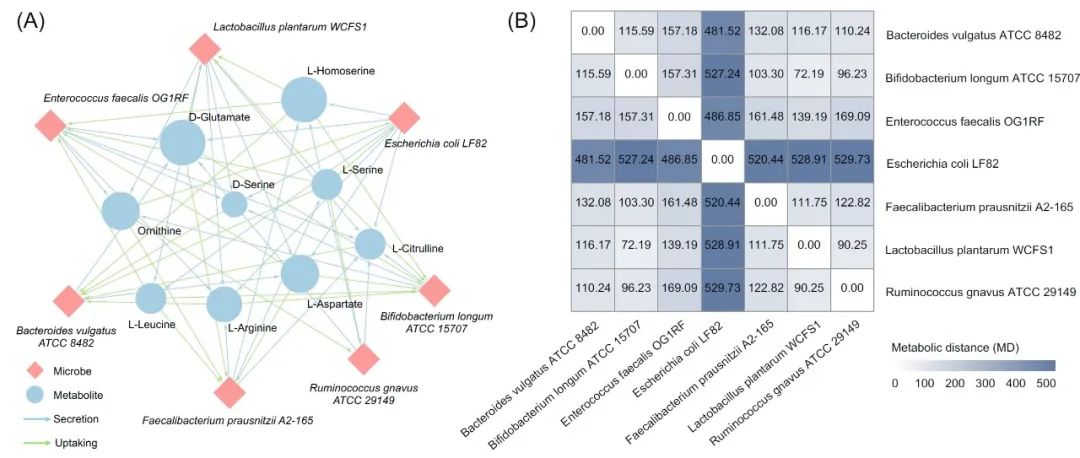
图2. 由PhyloMint PTM生成的为微生物-代谢物二分网络和由iNAP2.0生成的代谢距离热图
(A)iNAP2.0输出的微生物-代谢物(潜在可转移代谢物)二分网络;(B)iNAP2.0生成的代谢距离热图。结果来自演示数据1。
常见报错与解决办法
报错1(可能出现在全部步骤):报错信息为“An error occurred while updating information with the server.”该报错信息说明服务器正处于高负载状态或正在维护。解决办法是等候或等待使用人数较少的时候再运行任务。
报错2(可能出现在步骤1、2、3):报错信息为“Error opening file: No such file or directory.”该报错原因可能有二,其一是文件的扩展名不正确,其二是所有输入文件未被直接压缩,而是在文件夹中被压缩。解决办法分别是修正扩展名和直接压缩所有序列文件。
报错3(可能出现在步骤1、2、3):报错信息为“Error: Sequence file number exceeds the limit of 300.”该报错原因是上传了超过300个基因组或蛋白质序列文件。由于运载iNAP2.0的服务器计算资源有限,我们目前设置了一个数量上限。解决办法是减少模型数量或使用其他计算资源本地化运行相应的程序(如SMETANA)。
报错4(可能出现在8、9、10、11):可能出现的问题是程序一直不结束。这可能是由于SMETANA程序的高耗时造成的。解决办法仍是减少模型数量。
代码和数据可用性
iNAP2.0平台已对所有研究者们公开,允许免费注册并使用(https://inap.denglab.org.cn)。演示数据集被存放于共享数据中。
引文格式:
Peng, Xi, Kai Feng, Xingsheng Yang, Qing He, Bo Zhao, Tong Li, Shang Wang, and Ye Deng. 2024. “ iNAP 2.0: Harnessing metabolic complementarity in microbial network analysis.” iMeta e235. https://doi.org/10.1002/imt2.235
作者简介

彭玺(第一作者)
● 中国科学院生态环境研究中心直博生,导师为邓晔研究员,2024年公派至比利时荷语鲁汶大学交换学习。
● 目前研究方向为微生物生态学与生物信息学,相关学术成果已发表于 Nature Communications、iMeta等期刊。

邓晔(通讯作者)
●中国科学院生态环境研究中心研究员,中国科学院大学资源与环境学院博士生导师。
● 中国科学院“百人计划”终期评估优秀,国家计划青年拔尖人才。研究工作围绕环境微生物群落生态学开展,通过一系列的分子检测工具和数据分析手段的开发,对全球气候变化下微生物的响应和反馈机制以及微生物的地理学分布格局开展了一系列创新性的工作。《The ISME Journal》资深主编,《Microbiome》等五本SCI期刊副主编。2018-2023年连续六年获科睿唯安(Clarivate)全球高被引科学家,入选斯坦福大学全球2%高影响力学者和爱思唯尔(Elsevier)中国高被引学者,迄今在各类杂志上已发表SCI论文200多篇,文章总引用25000余次,H-index为78。

冯凯(通讯作者)
● 中国科学院生态环境研究中心助理研究员,iMeta青年编委。
● 从事微生物生态学和生物信息学研究,主要关注复杂生物群落的种间互作模式、微生物群落构建机制和气候变化下的微生物群落响应与生态效应。以第一和共同第一作者在One Earth, Science Bulletin,iMeta, Molecular Ecology Resources, Molecular Ecology等期刊发表多篇学术论文。
更多推荐
(▼ 点击跳转)
iMeta | 引用14000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底

3卷4期

3卷4期封底

1卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 2318
2318

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









