






研究论文
● 期刊:Cell Host & Microbe(IF:20.6)
● DOI:https://doi.org/10.1016/j.chom.2023.10.011
●原文链接: https://www.sciencedirect.com/science/article/pii/S1931312823004158
● 第一作者:张程程
● 通讯作者:翟齐啸(zhaiqixiao@sina.com)& 张家超(jiachao@hainanu.edu.cn) & 黄适(shihuang@hku.hk)
● 发表日期:2023-11-21
● 主要单位:
江南大学食品学院、海南大学食品科学与工程学院、香港大学牙医学院、江南大学食品科学与资源国家重点实验室、江南大学附属医院
摘要Summary
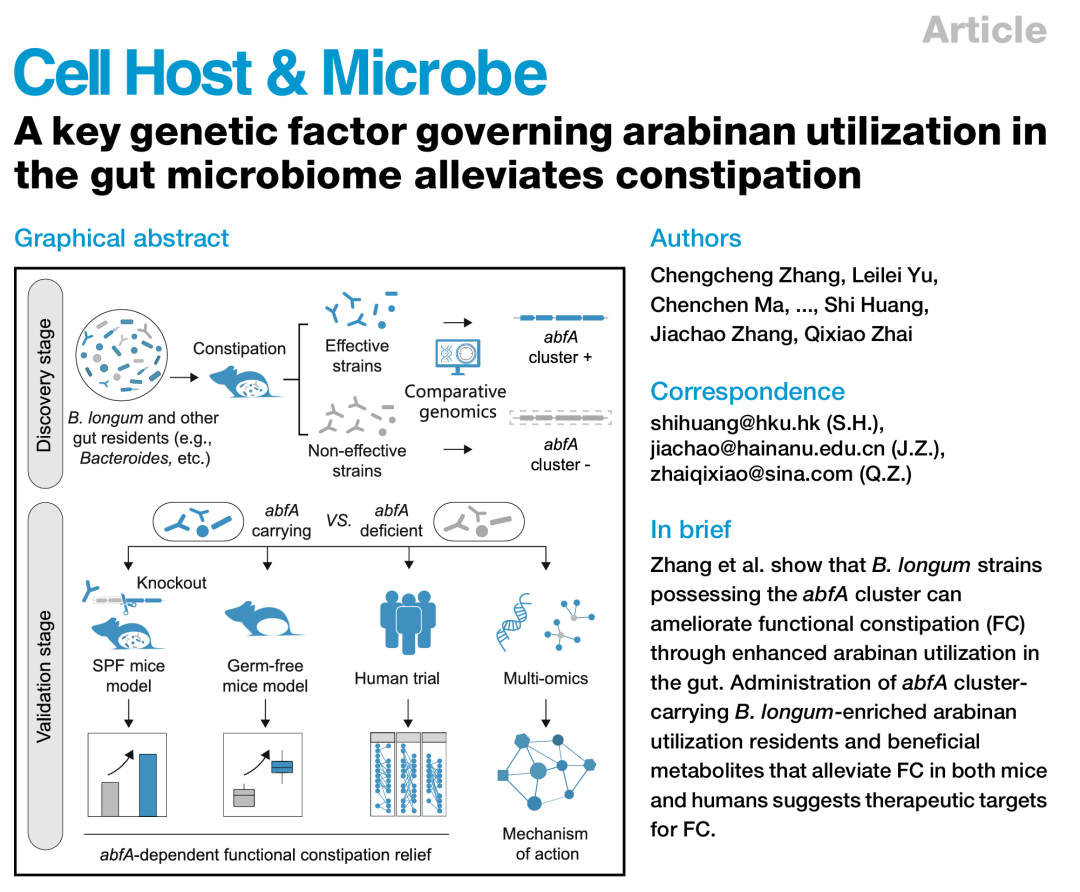
胃肠道蠕动障碍与肠道菌群失调相关。益生菌,如长双歧杆菌,可以改善这种肠道疾病;然而,此疗效依赖于菌株类型。我们发现,长双歧杆菌(Bifidobacterium longum)中的遗传因子abfA基因簇在阿拉伯糖利用过程中,影响长双歧杆菌对功能性便秘(functional constipation, FC)的治疗效果。在FC小鼠中,长双歧杆菌菌株(但不是abfA突变株)改善了胃肠道蠕动状况,而这种效果依赖于饮食中的阿拉伯糖。我们还发现,其他共生细菌中也存在abfA基因,其在改善小鼠FC中的作用也同样依赖于abfA基因。通过一项双盲、随机、安慰剂对照的临床试验,补充携带abfA基因簇的长双歧杆菌菌株(而非缺失abfA基因的菌株)能够富集阿拉伯糖利用菌群,增加有益代谢物,并改善FC症状。在多个人群实验中,abfA基因簇的丰度可以预测FC,而将富含abfA基因簇的人的肠道菌群移植到FC诱导的无菌小鼠体内,改善了肠道蠕动。总的来说,这些研究结果表明微生物abfA基因簇在改善FC中的作用,并为基因组定向益生菌治疗提供了原则依据。
引言Introduction
功能性便秘(FC)是全球人群中广泛存在的肠道疾病,全球患病率为10.1%–15.3%。功能性便秘主要表现为反复性排便不频、排便困难或排便不完全,并与心血管疾病、帕金森病、精神障碍、儿童自闭症和结直肠癌等相关联。
胃肠道蠕动障碍常与肠道菌群失调相关,表现为有益微生物(如双歧杆菌、乳酸嗜酸乳杆菌和植物乳杆菌等)丰度显著下降。这些微生物传统上被视为益生菌。为缓解功能性便秘,口服益生菌已被广泛应用。我们此前对15项随机对照临床试验的荟萃分析显示,益生菌的使用可使排便频率每周增加0.98次(95%置信区间[CI]:[0.36, 1.60]),并将全肠道通过时间缩短13.75小时(95% CI:[5.56, 21.93])。此外,益生菌摄入能够调节肠道微生物发酵的终产物(如短链脂肪酸,SCFAs),这些代谢产物通过与宿主免疫系统和肠神经系统的相互作用,有助于改善胃肠道蠕动功能。
然而,益生菌对功能性便秘(FC)的治疗效果在同一物种的不同菌株之间常常存在显著差异。由于机制不明,合理选择益生菌对医疗专业人员和患者来说仍然具有挑战性。普遍认为,细微的基因变异往往会导致益生菌在体内表现出不同的功能特性。因此,识别那些对功能性益生菌菌株特有的关键遗传元素,无论其是否存在,具有重要的科学和临床研究意义。然而,进行这一研究所需的实验量令人望而却步。(1)为了在动物模型中重现菌株特异性问题,研究实验室必须建立一个足够大的益生菌菌株库,并配套基因组数据,探索遗传因素与体内功能差异之间的因果关系。(2)在动物和人类模型中建立这种基因型与表型之间的联系更为关键。关于益生菌对胃肠道蠕动有益作用的大部分证据主要来源于小鼠模型的研究。益生菌菌株在动物模型中通常有效,但在人体临床试验中常常失败,或在人类中验证不充分。因此,基于人群队列的概念验证研究,以及结合体外和动物研究的证据,急需用于转化研究。
本研究旨在识别并系统验证外源益生菌或常驻肠道菌群影响胃肠道蠕动的关键遗传因子。首先,在一系列野生双歧杆菌(B. longum)菌株的综合库中,我们识别出了一个关键遗传因子(abfA基因簇,优先增强阿拉伯糖的利用),该因子在动物模型中有效缓解了FC,并通过基因敲除实验进一步验证了其功能作用。接下来,通过一项嵌套病例对照临床试验和人体粪便菌群移植(fecal microbiota transplantation, FMT)实验,结合宏基因组学和代谢组学,进一步确认了其在改善人类FC中的功能作用。值得注意的是,除了长双歧杆菌外,abfA基因/基因簇在肠道常驻微生物中也普遍存在,促进阿拉伯糖在肠道菌群中的利用,并改善小鼠和人类的便秘相关症状(图1A)。综上所述,我们强调了识别和表征决定益生菌在胃肠道疾病治疗效果差异的关键遗传因子的重要性和必要性。
结果Results
长双歧杆菌在小鼠中对洛哌丁胺引起的FC表现出菌株特异性的效应
从354名居住在中国17个省份或直辖市的中国人中分离出了185株长双歧杆菌菌株,年龄范围从0到108岁(数据S1)。我们观察到这些野生菌株之间具有较高的基因组广泛相似性(平均核苷酸同一性[ANI]范围:98.02%–100%)(图S1A)。基于这821个核心基因的最大似然法系统发育分析将这185株长双歧杆菌菌株分为5个不同的谱系(图1B;数据S1)。接下来,我们测试了这五个谱系代表性长双歧杆菌菌株在功能性上的差异,即它们对洛哌丁胺诱导的便秘(FC)在特定病原体无菌(SPF)BALB/c小鼠模型中的菌株特异性治疗效应(图1B和1C)。口服给药B. lon_4、B. lon_70和B. lon_8可增加粪便频率、胃肠道通过率、粪便水分含量,并减少全肠道通过时间(单因素方差分析,p < 0.01;图1D–1G)。相比之下,B. lon_39和B. lon_6未能缓解小鼠的FC症状(单因素方差分析,p > 0.05;图1D–1G)。值得注意的是,接受有效菌株移植的小鼠粪便中醋酸盐的含量更高(图1H–1J)。总体而言,我们捕捉到了长双歧杆菌在小鼠FC治疗效能中的菌株特异性。
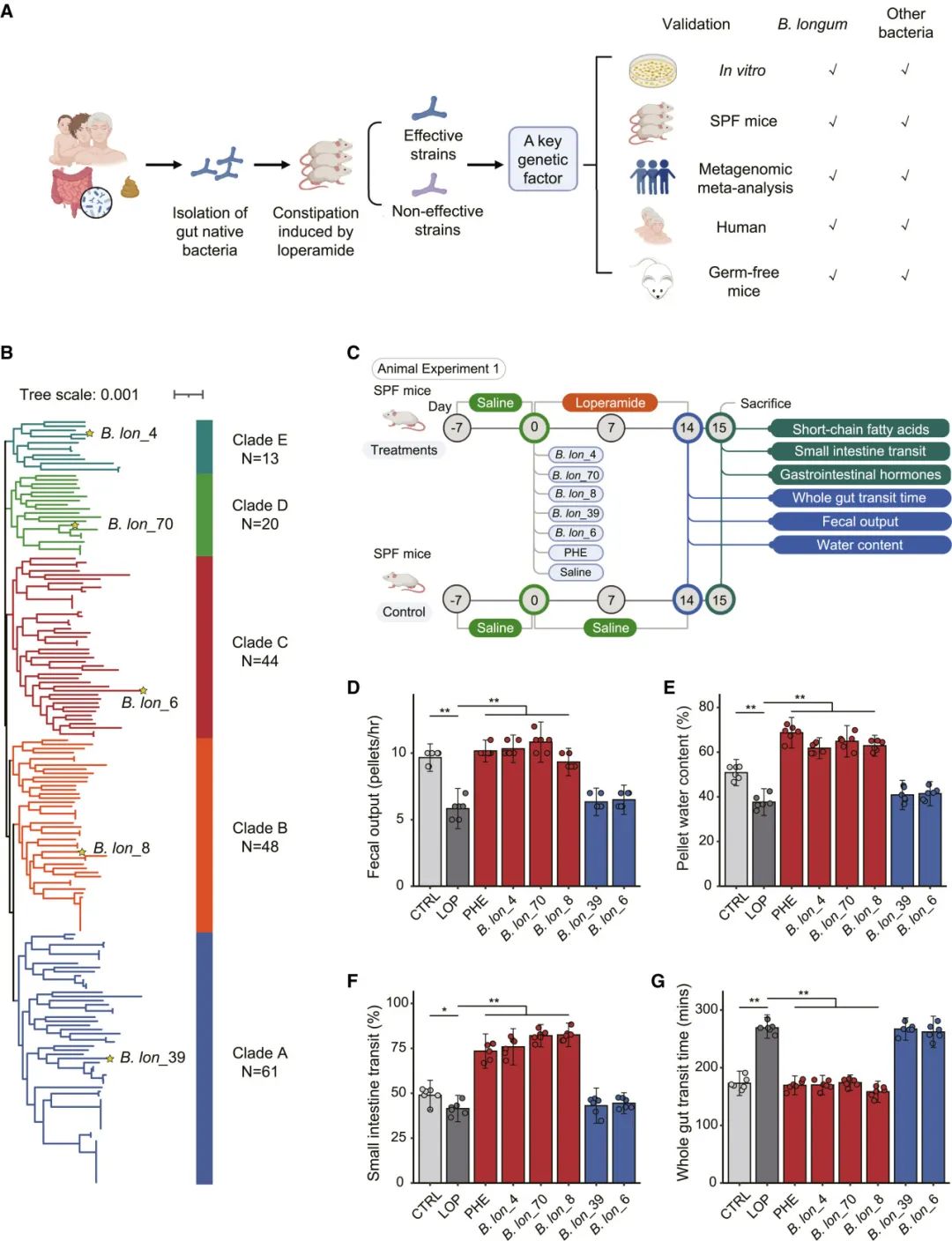
图1 | 长双歧杆菌在小鼠洛哌丁胺诱导的功能性便秘中表现出显著的菌株特异性效应
(A) 以功能基因组学为导向的益生菌筛选策略,用于治疗功能性便秘(FC)。(B) 185株野生长双歧杆菌菌株的全基因组系统发育树,其中识别出5株代表性菌株。(C) 实验设计。(D–G) 五株代表性菌株对小鼠功能性便秘(由洛哌丁胺诱导)的影响:(D) 粪便排出量,(E) 粪球水分含量,(F) 小肠蠕动时间,(G) 整个肠道通过时间。条形图表示平均值 ± 标准误(n = 6)。CTRL,控制组;LOP,洛哌丁胺;PHE,酚酞。(H–J) 五株长双歧杆菌菌株对粪便中 (H) 醋酸,(I) 丙酸,(J) 丁酸浓度的影响。
在所有箱线图中,中央线表示中位数,箱体的边缘显示第25和第75百分位数,须状线延伸至1.5倍的四分位距(n = 6)。∗p < 0.05,∗∗p < 0.01,采用单因素方差分析(ANOVA)确定。(A)和(C)是使用BioRender.com创建的。
有效的长双歧杆菌菌株携带两个特定基因位点(abfA和hypBA),编码阿拉伯呋喃糖苷酶
为了深入了解益生菌的有效性,我们对这些有效和无效菌株进行了比较基因组分析。菌株间的ANI范围较窄(即98.4%–99.1%),表明菌株间的功能差异不一定与益生菌基因组中核苷酸的全局模式相关,而可能仅由1%的遗传差异引起。接下来,我们使用OrthoMCL v2.0.9软件(默认参数)识别出了在有效长双歧杆菌菌株中存在但在无效菌株中缺失的五个同源基因位点(数据S1)。在位点1(以下简称abfA位点)处,我们发现了一个有效菌株特异性的abfA基因簇(以下简称abfA簇),该簇包含四个相连的基因,所有基因的功能注释均为α-L-阿拉伯呋喃糖苷酶(abfA)。在位点4(以下简称hypBA位点)处,我们还发现了一个有效菌株特异性的hypBA基因簇(hypBA簇),其中包括六个基因,分别编码β-半乳糖苷酶、β-L-阿拉伯二糖苷酶、β-L-阿拉伯呋喃糖苷酶和三个ATP结合盒(ABC)转运蛋白通道(图2A和2B)。在另外三个位点(位点2、3和5)中,非有效长双歧杆菌基因组中也存在平行基因,因此未在本研究中包含(数据S1)。有趣的是,这两个基因簇与降解膳食阿拉伯甘露糖(阿拉伯糖)功能相关,阿拉伯甘露糖是多种植物组织中果胶多糖的重要组成部分。
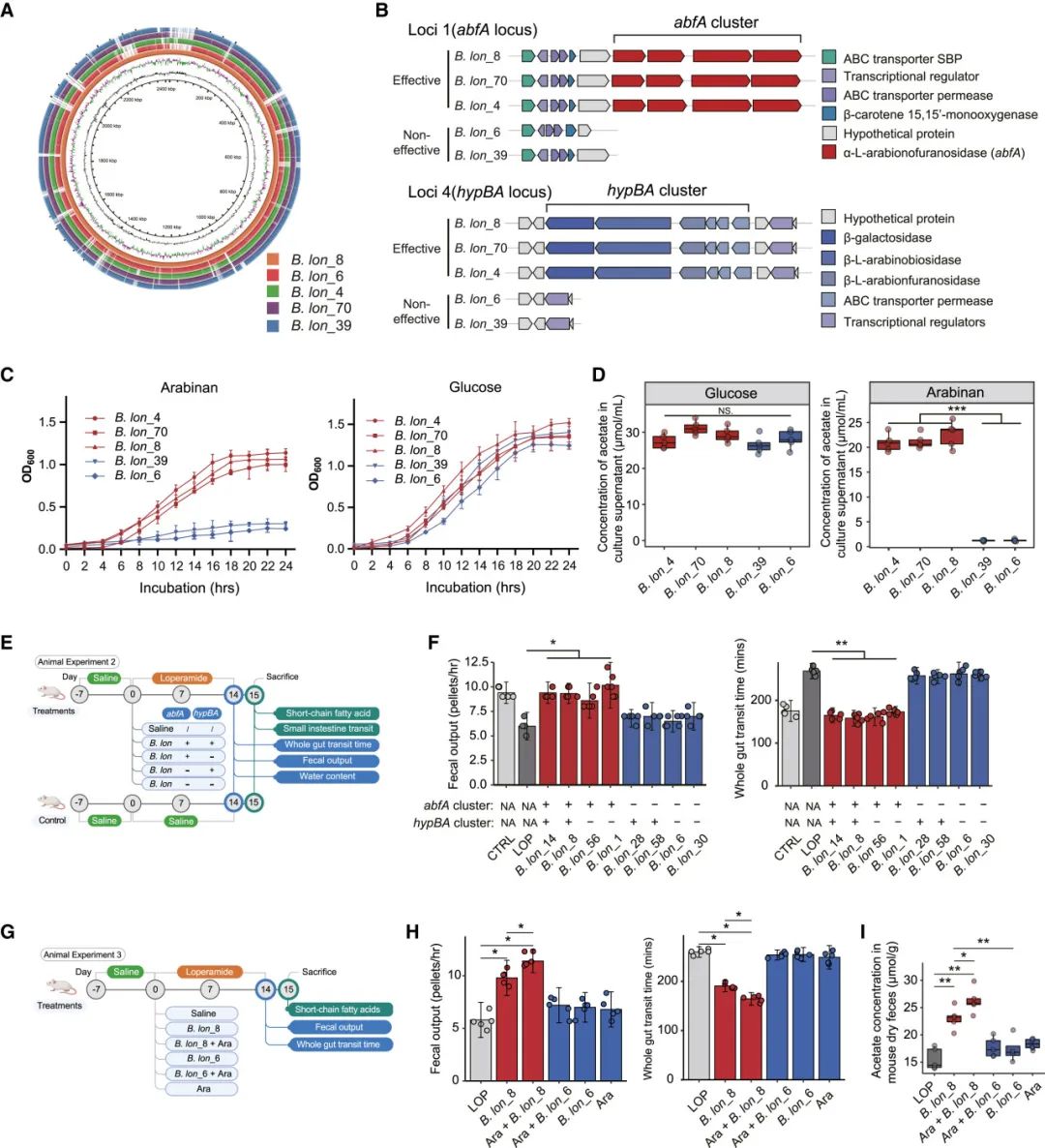
图2 | abfA簇是长双歧杆菌介导的便秘缓解的关键功能元件
(A) 有效和无效菌株染色体的基因组比较。从内到外依次为:(1)GC含量,(2)GC倾斜度,以及(3)五个菌株。(B) 长双歧杆菌菌株的abfA和hypBA位点的基因组结构。基因及其方向用箭头表示。(C) 有效和无效长双歧杆菌菌株在含葡萄糖或阿拉伯甘露糖的培养基中的生长曲线。误差条表示±标准误(n = 5)。(D) 箱线图显示在加入葡萄糖或阿拉伯甘露糖作为底物后的24小时培养液中醋酸盐的浓度。(E) 实验设计。(F) 采用含有abfA簇、hypBA簇或两者的八株长双歧杆菌菌株治疗FC小鼠后症状变化。(G) 实验设计。(H) abfA簇和膳食阿拉伯甘露糖在改善FC小鼠症状中的协同作用。(I) 治疗后第14天的粪便醋酸盐浓度,通过气相色谱/质谱法(GC/MS)测定。在(D)、(I)、(F)和(H)中,∗p < 0.05,∗∗p < 0.01,采用单因素方差分析(ANOVA)确定。(E)和(G)是使用BioRender.com创建的。
接下来,我们将这五株菌株返回到体外条件下,测试这种菌株水平的遗传差异是否导致不同的阿拉伯甘露糖利用能力。在含阿拉伯甘露糖的培养基中,三株有效菌株在24小时培养中生长速度明显快于无效菌株(图2C)。此外,经过阿拉伯甘露糖高效利用后,有效菌株显著增加了醋酸的产生(>20 μmol/mL),这是益生菌双歧杆菌在碳水化合物合成和代谢中的关键终产物,而无效菌株则未能产生有意义水平的醋酸(<1 μmol/mL;图2D)。相比之下,在含葡萄糖的培养基中,我们没有观察到菌株之间的这种表型差异(图2C和2D)。接下来,我们检查了在阿拉伯甘露糖或葡萄糖培养下,这两个位点(即abfA和hypBA位点)中每个基因的体外转录水平活性。已有研究报告称,阿拉伯甘露糖能够提高长双歧杆菌 JCM1217中类似abfA位点的BLLJ_1852和BLLJ_1853基因的表达水平。27类似地,我们发现阿拉伯甘露糖上调了B. lon_8中abfA位点的十个基因中的八个(BLLJ_1850、BLLJ_1852、BLLJ_1853等),其中三个基因(即BLLJ_1850、BLLJ_1852和BLLJ_1853)属于abfA簇(图S1B)。相比之下,hypBA位点中的十一基因中只有三个基因的表达略有上调,其中两个基因属于hypBA簇。在abfA位点,相对mRNA表达量与16S rRNA基因的比值(%)平均为35%,而在hypBA位点则仅为1.9%(图S1B)。值得注意的是,abfA簇中的两个基因(即BLLJ_1852和BLLJ_1853)相对于16S rRNA的表达水平超过60%,这表明它们在阿拉伯甘露糖降解中的主导功能作用。考虑到基因表达的水平和广度,abfA位点(或abfA簇)对阿拉伯甘露糖利用的贡献远大于hypBA位点。根据体外细菌生长实验、代谢组学分析、基因组学分析和mRNA表达谱的证据,三株有效长双歧杆菌菌株的益生特性主要归因于abfA簇的积极参与,促进了阿拉伯甘露糖的高效利用。
abfA簇是长双歧杆菌介导的便秘缓解的关键因素
接下来,我们测试了长双歧杆菌基因组中的abfA簇或hypBA簇是否能够独立影响小鼠便秘(FC)治疗效果,使用我们菌株库中的野生菌株。从最初的185株长双歧杆菌菌株中,105株(56.8%)携带完整的abfA簇,而129株(69.7%)携带完整的hypBA簇。有趣的是,185株菌株中有79株(42.7%)同时携带这两个基因簇(图S1C;数据S1)。从这个库中,选择了八株长双歧杆菌菌株,这些菌株在其基因组中具有不同的abfA簇和hypBA簇的存在/缺失模式,用于体外和动物实验(图2E)。首先,使用阿拉伯甘露糖作为唯一碳源的体外培养实验表明,携带abfA簇的菌株(abfA+hypBA−或abfA+hypBA+)在6小时培养后生长速度显著快于其他菌株(图S1D和S1E)。接下来,携带abfA簇的菌株(abfA+)持续增加了粪便排放量并减少了全肠道通过时间(p < 0.05;图2F),而仅编码hypBA簇的菌株(abfA−hypBA+)对小鼠便秘症状的逆转没有影响(p > 0.05;图2F)。结合额外的验证实验(图S1F–S1J),我们推测abfA簇可能是决定益生菌效能的关键遗传因素,其中不涉及洛哌丁胺的直接微生物代谢(图S1K和S1L)。
鉴于abfA簇专门用于阿拉伯甘露糖的利用,我们接下来研究了有效长双歧杆菌菌株与膳食阿拉伯甘露糖在FC小鼠中的协同作用(图2G)。首先,单独的阿拉伯甘露糖处理未影响粪便排放量或全肠道通过时间(图2H)。此外,补充阿拉伯甘露糖与携带abfA簇的B. lon_8相比单独使用B. lon_8显著增加了粪便排放量并减少了全肠道通过时间(图2H)。相比之下,即使添加了足够的膳食阿拉伯甘露糖,缺乏abfA簇的B. lon_6对FC症状没有影响(图2H)。尽管补充B. lon_8显著增加了肠道中的醋酸生成,但膳食阿拉伯甘露糖与B. lon_8的联合补充可以进一步促进肠道醋酸的生成(图2I)。值得注意的是,考虑到阿拉伯甘露糖可能存在于正常饮食中,我们进一步在阿拉伯甘露糖去除饮食的小鼠中重复了上述实验(图S1M–S1O)。单独给药B. lon_8未能改善FC症状,进一步证明了携带abfA簇的长双歧杆菌菌株与阿拉伯甘露糖在小鼠FC中的协同作用。
证据表明长双歧杆菌的abfA簇在增强阿拉伯甘露糖利用和改善便秘(FC)中的因果作用
有文献记录表明,阿拉伯糖是阿拉伯甘露糖的降解产物,可能会阻碍蔗糖的消化,从而导致蔗糖异麦芽糖酶缺乏和腹泻,这也有助于改善便秘(FC)。29,30 在动物实验4和体外实验中(图S2A–S2H),我们确认肠道菌群中的abfA簇在降解阿拉伯甘露糖并生成阿拉伯糖的过程中发挥了关键作用,这一过程发生在结肠中,而非在小肠空肠中(蔗糖消化的部位)。因此,B. lon_8对FC的缓解并非由阿拉伯甘露糖降解生成的阿拉伯糖通过阻断蔗糖代谢所致。
在远端结肠中,微生物代谢果糖会产生大量的醋酸,这可能与abfA簇相关的醋酸生产有关。我们的体外实验(图S2I–S2K)表明,B. lon_8的加入对小鼠肠道菌群(MGM)利用果糖生成醋酸没有显著影响,但对MGM通过降解阿拉伯甘露糖生成醋酸的能力有积极影响。这些结果排除了B. lon_8通过果糖-醋酸轴发挥作用的假设。
接下来,我们探索了携带abfA簇的长双歧杆菌给药、FC症状变化、果糖消耗、阿拉伯糖生成和醋酸生成之间的因果关系(图3A)。FC症状指标(如粪便排放量和全肠道通过时间)的时间变化与B. lon_8治疗的存在/缺失模式波动高度一致,这与醋酸和阿拉伯糖浓度的波动相符(图3B)。相比之下,粪便中的果糖浓度在整个实验过程中高度稳定,无论是FC诱导还是益生菌补充(图3B)。这项体内纵向研究进一步提示,长双歧杆菌的abfA簇在增强肠道中未充分利用的阿拉伯甘露糖的利用和改善便秘(FC)症状中起着因果作用。
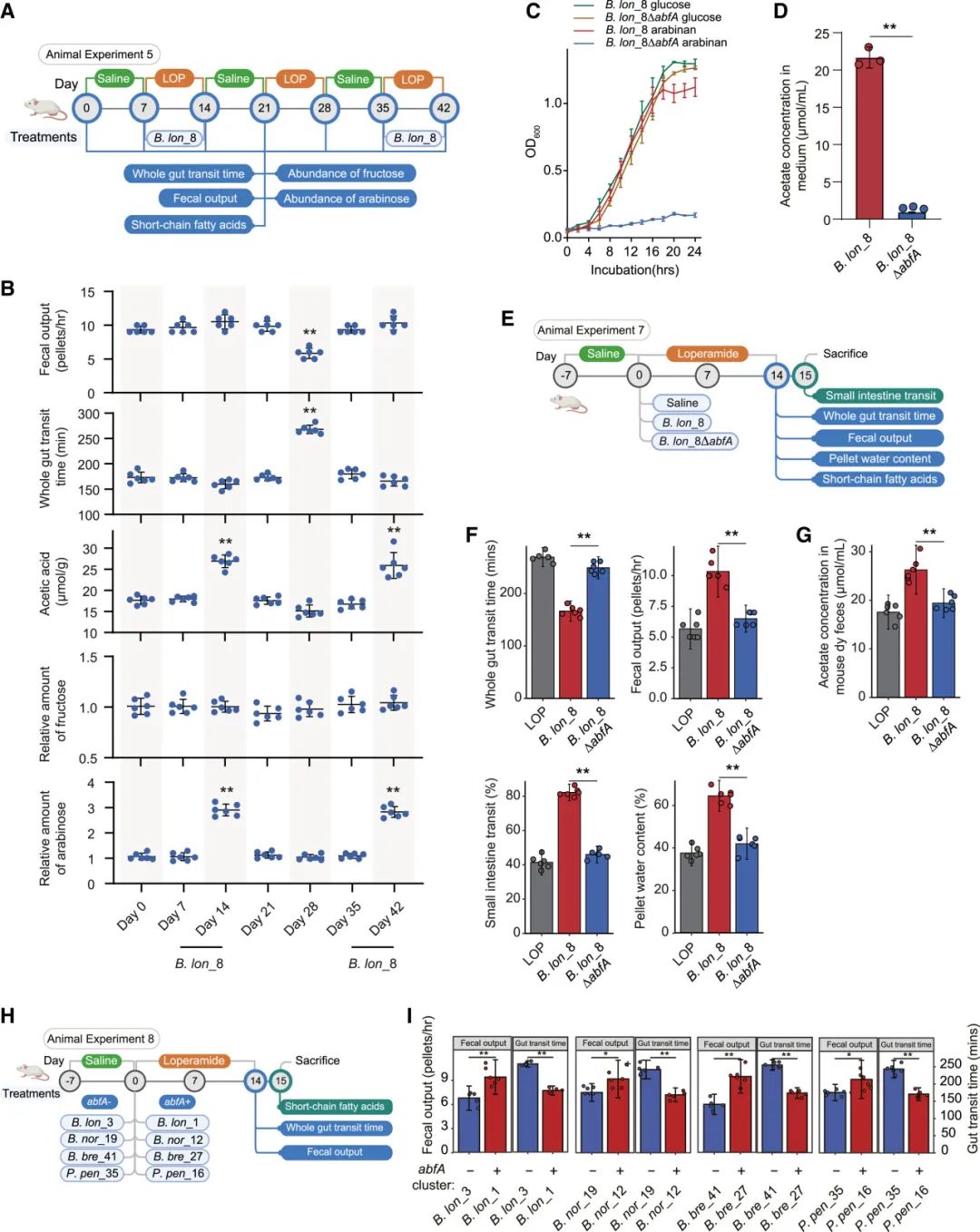
图3 | 长双歧杆菌的abfA簇在小鼠便秘(FC)改善中的因果作用
(A) 实验设计。
(B) 携带abfA簇的长双歧杆菌菌株对FC症状、醋酸浓度以及小鼠粪便中果糖或阿拉伯糖相对含量的影响,时间跨度为42天。果糖和阿拉伯糖的量相对于第0天的值进行显示。蓝色点代表单个小鼠。
(C) B. lon_8和B. lon_8 ΔabfA在含葡萄糖或阿拉伯甘露糖培养基中的生长曲线(n = 3/组)。
(D) B. lon_8和B. lon_8 ΔabfA在含葡萄糖或阿拉伯甘露糖的培养基中经过24小时体外培养后的醋酸浓度。
(E) 实验设计。
(F) B. lon_8和B. lon_8 ΔabfA对小鼠FC改善的影响。
(G) B. lon_8和B. lon_8 ΔabfA对小鼠便秘(FC)症状下粪便中醋酸浓度的影响。
(H) 实验设计。
(I) abfA簇对非长双歧杆菌菌株改善小鼠FC的影响。
∗p < 0.05,∗∗p < 0.01,通过Wilcoxon秩和检验(D),t检验(I),或单因素方差分析(B, F, G)确定。在(D, F, G, 和 I)中,柱状图表示平均值 ± 标准误差(SEM)。(A)、(E)和(H)通过BioRender.com制作。
我们通过扩展的泛基因组分析和功能缺失方法进一步验证了其因果作用。首先,我们在15个野生株的长双歧杆菌中识别了4,648个基因,这些菌株中有8个是有效的,7个是无效的。在这4,648个基因中,只有4个注释为abfAs基因,且这些基因趋向于存在于有效株中,而有一个基因趋向于存在于无效株中(卡方检验,p < 0.05),而其他基因并不特定于有效或无效菌株(图S3A)。接下来,我们在B. lon_8中通过同源重组敲除了abfA簇中的两个关键基因(BLLJ_1852和BLLJ_1853),这两个基因主要通过细菌对阿拉伯甘露糖的利用而上调,生成了突变株:B. lon_8ΔabfA(图S3B和S3C)。B. lon_8ΔabfA在阿拉伯甘露糖培养基中的体外生长被显著抑制,且与B. lon_8相比,其醋酸的生成显著减少,但在葡萄糖培养基中未观察到这种菌株间的差异(图3C, 3D和S3D)。更重要的是,在B. lon_8ΔabfA处理的小鼠中,观察到对FC的保护效果远低于B. lon_8处理的小鼠,且小鼠的粪便中醋酸浓度较低(图3E–3G)。最后,我们还确认了携带abfA簇的长双歧杆菌菌株在FC或健康小鼠中均不引起腹泻(图S4A–S4F)。
abfA簇对于非长双歧杆菌肠道微生物在阿拉伯甘露糖利用和改善便秘(FC)中的能力也起着关键作用
我们进一步在NCBI数据库中全局性地调查了abfA簇或abfA基因及其所属的菌群体。值得注意的是,abfA同源基因广泛分布在189个细菌基因组中,涵盖50个物种和22个属(主要包括拟杆菌属、双歧杆菌属、肠球菌属、乳酸乳球菌属等)(数据S1)。根据江南大学的大型菌株库(食品微生物培养物收藏 [CCFM];STAR方法),我们识别了三种具有abfA簇/基因的人类肠道分离株(双歧双歧杆菌 [B. bre],乳酸乳球菌 [P. pen],拟杆菌Nordii [B. nor])及其野生型abfA缺失突变株。这六个菌株进一步用于治疗小鼠便秘(FC)(图3H)。与长双歧杆菌相关的实验一致,这些携带abfA基因的肠道共生菌显著增加了粪便排放量,并减少了全肠道通过时间,而缺失abfA簇的菌株未能改善FC症状(图3I)。尽管在携带abfA基因的菌株中观察到了相似的表型变化,但治疗后短链脂肪酸(SCFAs,包括醋酸、丙酸和丁酸)的增加在不同菌株间有所依赖:长双歧杆菌(醋酸)、B. breve(醋酸)、P. pen(丙酸和丁酸)和B. nor(醋酸和丁酸)(图S4G–S4I)。有趣的是,我们的体外培养实验进一步显示:(1)B. nor的abfA簇在单培养中增强了细菌对阿拉伯甘露糖的利用,(2)携带abfA基因的长双歧杆菌可以促进B. nor在共培养中的生长(图S4J–S4L)。
总体而言,abfA簇是促进阿拉伯甘露糖同化并有效增加小鼠肠道运动性的关键遗传决定因子。
验证携带abfA簇的长双歧杆菌在中国老年人群体中对便秘(FC)的保护作用
在一项双盲、随机、安慰剂对照的临床试验中,分别向被诊断为便秘(FC)的老年人群体中给予了两种携带abfA簇的长双歧杆菌菌株(B. lon_8和B. lon_1)以及一种缺失abfA簇的菌株(B. lon_6)(图4A;STAR方法)。与我们在动物实验中的观察结果类似,B. lon_1和B. lon_8的28天治疗分别使粪便频率平均增加了1.04次/周(95% CI: [0.62, 1.51];数据S2)和1.15次/周(95% CI: [0.66, 1.64];数据S2)。B. lon_1和B. lon_8分别使Cleveland临床便秘评分(CCCS)从基线到第28天平均降低了4.09分(95% CI: [2.13, 6.05];数据S2)和4.86分(95% CI: [3.00, 6.36];数据S2)。相比之下,安慰剂组和B. lon_6组未观察到显著差异(图4B和4C)。B. lon_1和B. lon_8的补充还显著降低了便秘症状的患者评估(PAC-SYM)(B. lon_1组为−0.40 ± 0.56,B. lon_8组为−0.45 ± 0.40;数据S2)和便秘生活质量的患者评估全球评分(PAC-QoL)(B. lon_1组为−0.42 ± 0.44,B. lon_8组为−0.37 ± 0.27;数据S2),这一变化持续到干预结束(即第28天)。在随访访问(第56天)时,尽管在第28天到第56天期间未再进行益生菌补充,但PAC-SYM和PAC-QoL仍显著降低(数据S2)。此外,在B. lon_1和B. lon_8治疗的28天期间,直肠、腹部、粪便及身体不适显著减少(临床特征见数据S2)。
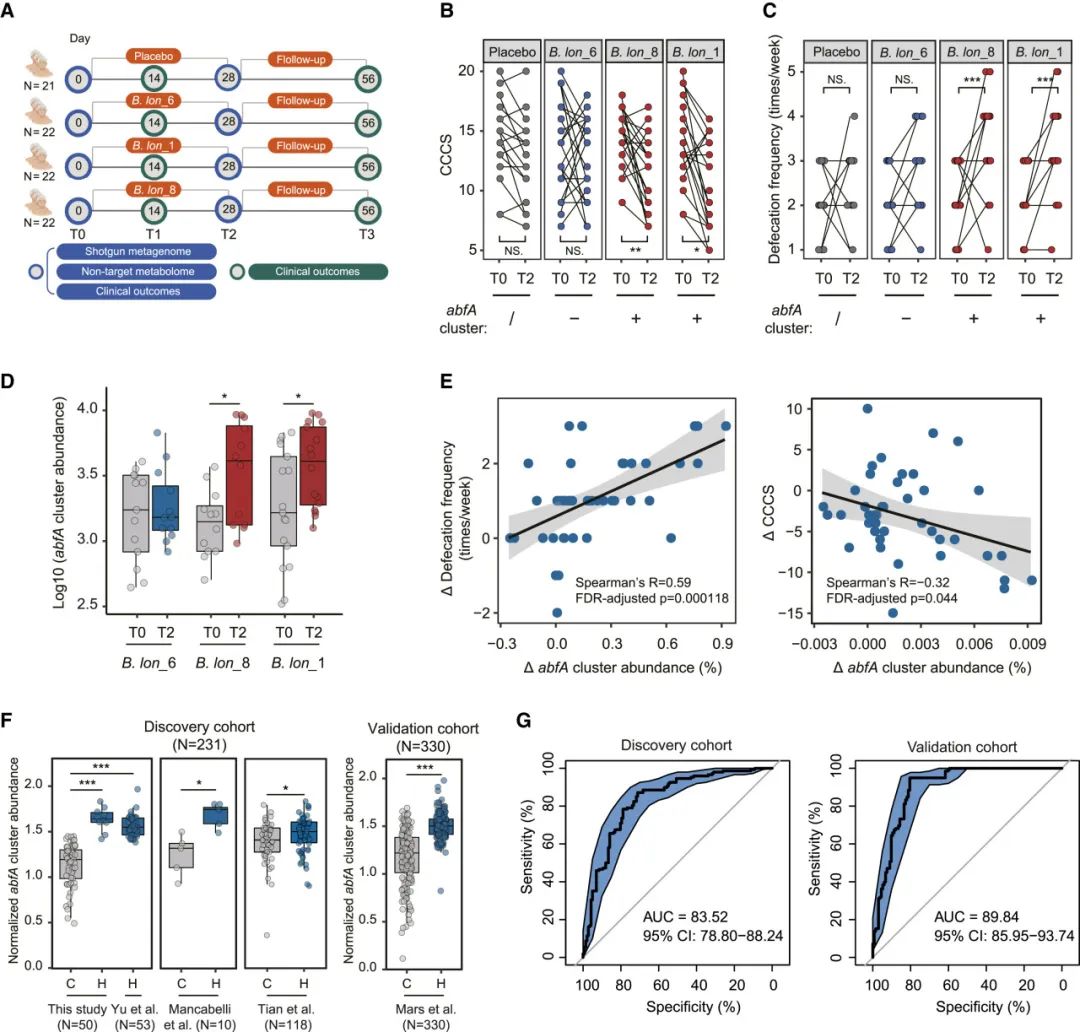
图4 | abfA簇在原生肠道菌群中对便秘(FC)的保护作用可以在多个人体队列中得到验证
(A) 实验设计。
(B 和 C) 28天补充含有abfA簇的长双歧杆菌菌株后,CCCS(B)和排便频率(C)的变化。安慰剂组,n = 21;B. lon_6组,n = 13;B. lon_8组,n = 12;B. lon_1组,n = 16。
(D) 28天补充长双歧杆菌菌株后,abfA簇的丰度增加。B. lon_6组,n = 13;B. lon_8组,n = 12;B. lon_1组,n = 16。
(E) 补充长双歧杆菌菌株后abfA簇丰度(从T0到T2)的时间变化与排便频率和CCCS显著相关。对所有便秘(FC)患者(n = 41)的Spearman相关性分析,分别针对B. lon_6组、B. lon_8组、B. lon_1组。
(F) 我们的元分析中人类受试者的标准化abfA簇丰度。
(G) 在发现队列和验证队列中,标准化abfA簇丰度的ROC分析。误差带表示95%置信区间。
在(B)–(D)和(F)中,∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,均通过配对学生t检验或Wilcoxon符号秩检验确定。(A)图由BioRender.com制作。
我们接着使用宏基因组学研究了有效的长双歧杆菌菌株是否以及如何影响粪便中微生物的abfA簇丰度。28天补充B. lon_1和B. lon_8显著增加了肠道菌群中abfA簇的丰度,而abfA簇缺失的B. lon_6则没有这样的效果(图4D)。相比之下,三种测试菌株的补充对肠道菌群中hypBA簇的丰度没有影响(图S5A)。abfA簇丰度的时间变化与排便频率(Spearman等级相关,r = 0.59,假发现率(FDR)调整后的p < 0.001;图4E)显著正相关,而与CCCS的变化则显著负相关(Spearman等级相关,r = −0.32,FDR调整后的p < 0.05;图4E)。然而,长双歧杆菌物种和hypBA簇的物种级丰度变化与CCCS和排便频率的变化没有相关性(Spearman等级相关,FDR调整后的p > 0.05;图S5B–S5E)。因此,我们推测abfA簇,而不是长双歧杆菌物种或hypBA簇,在防治便秘(FC)中发挥着关键的保护作用,并且其在健康与便秘之间的肠道菌群丰度差异可能是被之前的粪便宏基因组学研究忽视的。
全球多队列元分析揭示了abfA簇在预测人类肠道菌群中的便秘(FC)具有很强的预测能力
在这里,我们还分析了来自四个便秘(FC)队列的511个粪便宏基因组样本(n = 53,Coker 等人33;n = 10,Mancabelli 等人34;n = 118,Tian 等人35;和 n = 330,Mars 等人36),以验证我们的假设。与健康组相比,FC组的粪便宏基因组中abfA簇的丰度显著较低(Wilcoxon秩和检验,p < 0.05;图4F)。相比之下,hypBA簇的丰度在这些队列中的健康与FC之间没有显著差异(图S5F)。接下来,我们基于整个肠道群落水平上abfA簇的集合丰度(经测序深度标准化)构建了一个分类模型,用于区分健康与FC。该预测模型在发现队列中表现出较高的分类准确性(受试者工作特征曲线下面积 [AUC] = 83.52%,95%置信区间:78.80%–88.24%,n = 240;图4G),并且在外部队列(Mars 等人36,AUC = 89.84%,95%置信区间:85.95%–93.70%,n = 330;图4G)中得到了良好的验证。这表明,从宏基因组数据中估算的abfA簇丰度有可能发展成一种简单且有效的生物标志物,用于诊断便秘或预测便秘的治疗结果。
长双歧杆菌给药后,肠道微生物abfA簇丰度升高所引起的特定肠道常驻菌株的变化
在这里,我们首先测试了长双歧杆菌给药是否会引起肠道菌群落的结构变化。有趣的是,在属和种水平上,基于Bray-Curtis差异性分析,并没有观察到任何宿主组内肠道菌群落的差异(排列多元方差分析 [PERMANOVA],p > 0.05;图5A,S6A和S6B)。然而,通过28天给药B. lon_8和B. lon_1,我们观察到在菌株(基因组拼接图谱 [MAG])水平上,菌群落的组成发生了显著变化(PERMANOVA,B. lon_8组R2 = 0.0608,p < 0.05;B. lon_1组R2 = 0.0563,p < 0.05;图5A和S6C)。此外,abfA簇丰度的时间变化与基线和28天间的宿主内Bray-Curtis差异性在属水平(Spearman等级相关,r = −0.054,FDR调整后p = 0.71;图5B)或种水平(Spearman等级相关,r = 0.066,FDR调整后p = 0.71;图5C)没有相关性,而与菌株水平的差异性呈正相关(Spearman等级相关,r = 0.55,FDR调整后p = 0.00093;图5D)。
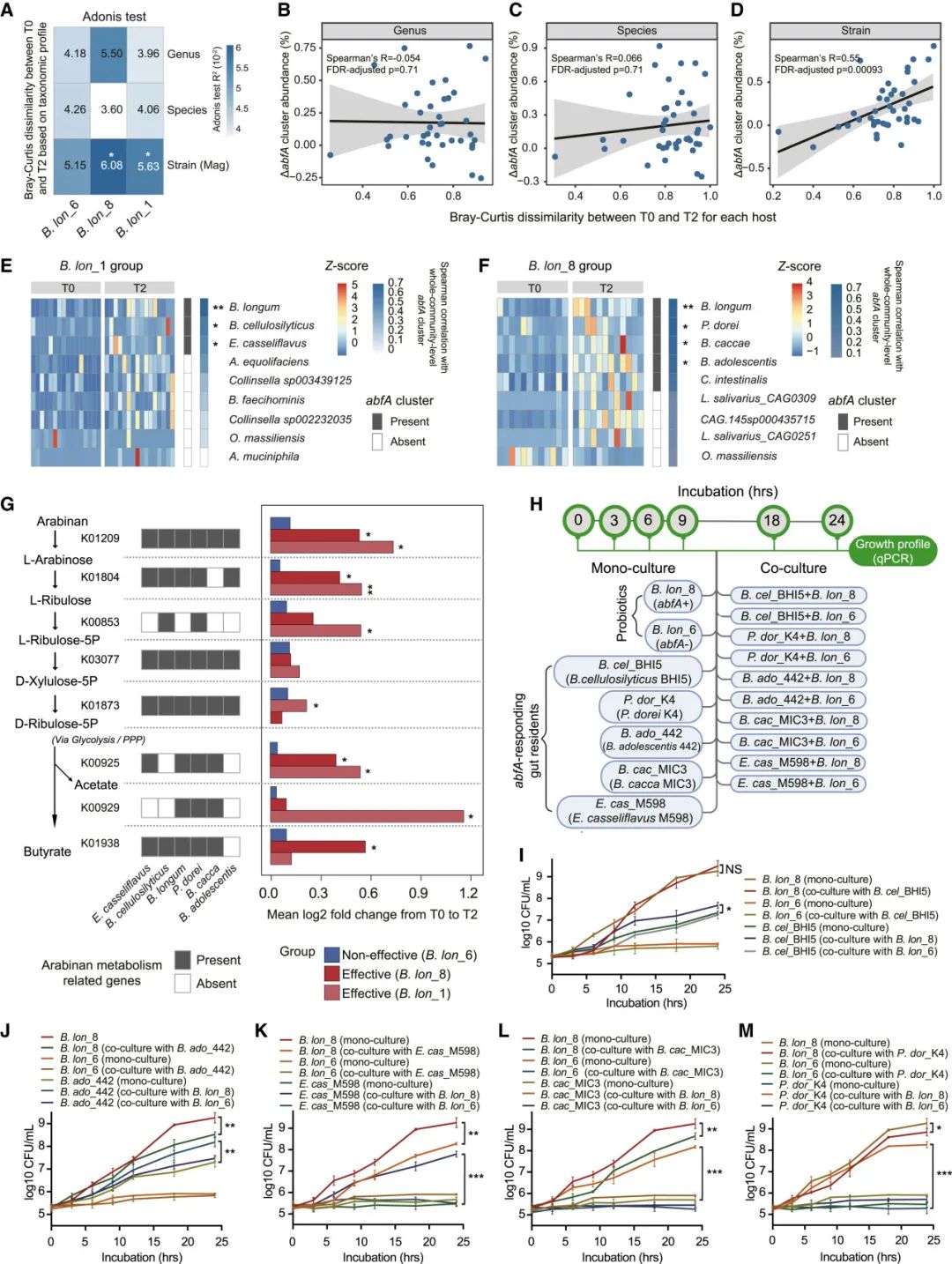
图5 | 由有效长双歧杆菌摄入后,肠道常驻菌群和功能基因对微生物abfA簇丰度升高的响应变化
(A) 由长双歧杆菌菌株在三种分类学水平上引起的T0和T2之间的人类肠道菌群差异。热图展示了Adonis测试产生的R2值。(B–D) abfA簇丰度的时间变化与T0和T2之间Bray-Curtis差异性之间的相关性,分别在(B) 属、(C) 种和(D) 菌株水平上。误差带表示95%的置信区间。(E和F) 28天给药后,(E) B. lon_1和(F) B. lon_8的标记MAGs。在左面板中,热图展示了每个受试者在零均值归一化(Z-score)下的MAG丰度。中间面板,热图显示了菌株是否携带任何abfA簇。右面板,展示了微生物菌株丰度变化与abfA簇丰度变化之间的Spearman相关性。符号表示显著性:∗p < 0.05,∗∗p < 0.01。(G) 肠道菌群中关键阿拉伯糖醛降解途径的功能基因与在长双歧杆菌植入后表现出丰度变化的肠道微生物之间的联系。在左面板中,展示了肠道菌群中阿拉伯糖醛利用的关键代谢途径。中间面板,热图显示了菌株是否携带与阿拉伯糖醛代谢相关的基因。右面板,柱状图展示了在有效(B. lon_1和B. lon_8)和非有效(B. lon_6)组中的阿拉伯糖醛代谢相关基因丰度的变化。(H) 实验设计。(I–M) (I) B. cel_BHI5、(J) P. dor_K4、(K) B. ado_442、(L) E. cas_M598和(M) B. cac_MIC3在以阿拉伯糖醛作为唯一碳源的培养基中,单独培养或与B. lon_8或B. lon_6共同培养时的生长曲线。(I–M) 误差条表示±标准误差(n = 3)。∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,采用单向ANOVA分析,24小时时的数据。(H)由BioRender.com创建。
我们接下来识别了对益生菌植入反应的肠道微生物,重点关注MAGs(n = 154)。同样,我们未能识别出与有效长双歧杆菌给药相关的任何常驻细菌属和物种(图S6A和S6B;数据S3),这与许多其他研究的结果大致一致。37,38 相反,17个常驻菌株(MAGs)在28天的B. lon_1或B. lon_8给药后,丰度发生了显著变化(Wilcoxon符号秩检验,p < 0.05;图5E和5F;数据S3)。其中有8个响应菌株在其基因组中携带abfA簇。在这8个菌株中,3个(长双歧杆菌、B. cellulosilyticus和E. casseliflavus)在B. lon_1治疗组的受试者中被识别,而5个(长双歧杆菌、P. dorei、B. caccae、B. adolescentis和C. intestinalis)在B. lon_8组被识别(图5E、5F、S6D和S6E)。尽管给药非有效菌株(B. lon_6)或安慰剂也引起了5个或4个肠道常驻菌丰度的时间变化,但它们与有效菌株治疗组的肠道微生物标志物没有重叠(图S6F和S6G)。接下来,我们对每个组内进行相关性分析,发现这8个携带abfA簇的菌株(除了B. lon_8组中的C. intestinalis)与B. lon_8或B. lon_1组中的全群体水平abfA簇丰度呈正相关(Spearman秩相关,FDR调整后的p < 0.05;图5E和5F;数据S3)。然而,在B. lon_6或安慰剂组中未发现这种关联(Spearman秩相关,FDR调整后的p > 0.05;图S6H和S6I;数据S3)。这表明,益生菌长双歧杆菌(abfA+)的给药促进了能够优先利用阿拉伯糖醛的肠道常驻菌群的群体结构变化。根据宏基因组数据,我们分析了给药后益生菌在粪便中的丰度,发现无论其对FC的有效性如何,三种菌株在第28天的丰度均未观察到显著差异(图S7A)。此外,在另一项动物实验中(图S7B),我们确认有效的长双歧杆菌菌株(n = 2)在小鼠的粪便和结肠内容物中的丰度与非有效的长双歧杆菌菌株(n = 2)相同(基于qPCR结果,图S7C和S7D),这与宏基因组结果大致一致。这些结果表明,菌株间的功能差异并不依赖于细菌丰度,而是取决于是否携带abfA簇。
我们接下来测试了这些关键菌株丰度变化是否富集了与阿拉伯糖降解相关的功能基因(KOs)。在B. lon_1或B. lon_8组中,总共有五个KOs与L-阿拉伯糖的发酵过程以及短链脂肪酸(SCFAs)的产生相关,在28天干预结束时显著升高(Wilcoxon符号秩检验,离散FDR调整后的p < 0.1;图5G;数据S4)。此外,这些关键常驻菌株至少携带了阿拉伯糖代谢中的八个基因(KOs)中的四个。
接下来,我们通过体外实验(图5H)测试了长双歧杆菌菌株是否以及如何诱导携带abfA簇的本地肠道菌群的生长(图5G)。我们将B. lon_8或B. lon_6与五种肠道微生物分离株共同培养(即B. adolescentis菌株442 [B. ado_442]、E. casseliflavus菌株M598 [E. cas_M598]、B. cellulosilyticus菌株BHI5 [B. cel_BHI5]、P. dorei菌株K4 [P. dor_K4]和B. cacca菌株MIC3 [B. cac_MIC3]),这些菌株与宏基因组中的菌株高度相似(图5H)。B. lon_8显著促进了B. cac_MIC3、P. dor_K4、E. cas_M598、B. cel_BHI5和B. ado_442的生长,而B. lon_6未能做到这一点(图5I和5M)。这些结果表明,携带abfA簇的长双歧杆菌可能为受体细菌提供阿拉伯糖降解产物(如L-阿拉伯糖)作为生长底物。
abfA簇丰度的升高改变了肠道菌群的代谢功能
接下来,我们通过28天的益生菌治疗,调查了粪便宏基因组和代谢组的变化。首先,发现有14个KOs的丰度在B. lon_1和B. lon_8处理后发生了变化(Wilcoxon符号秩检验,dFDR调整后的p < 0.1;图6A;数据S4),而在B. lon_6组中则保持不变。这些对处理响应的KOs涉及阿拉伯糖代谢、乙酸生产、甘露酸代谢、鞘胆酸生产和精胺生产(图6A;数据S4)。有趣的是,K10012(arnC,L-阿拉伯糖转移酶)、K02904(rpmC,大亚基核糖体蛋白)、K01804(araA,L-阿拉伯糖异构酶)、K01442(cbh,胆硫甘氨酸水解酶)、K01209(abfA,α-L-阿拉伯呋喃糖苷酶)、K00925(ackA,乙酸激酶)等KOs的丰度变化与整体abfA簇丰度的变化高度一致(Spearman秩相关,r > 0.3,FDR调整后的p < 0.05;图6A)。相反,我们还探讨了哪些常驻微生物主要贡献了肠道中响应性功能基因(KO)的整体丰度。预期中,例如,ackA与长双歧杆菌和B. adolescentis的丰度呈正相关(Spearman秩相关,r > 0.3,FDR调整后的p < 0.05;数据S4)。
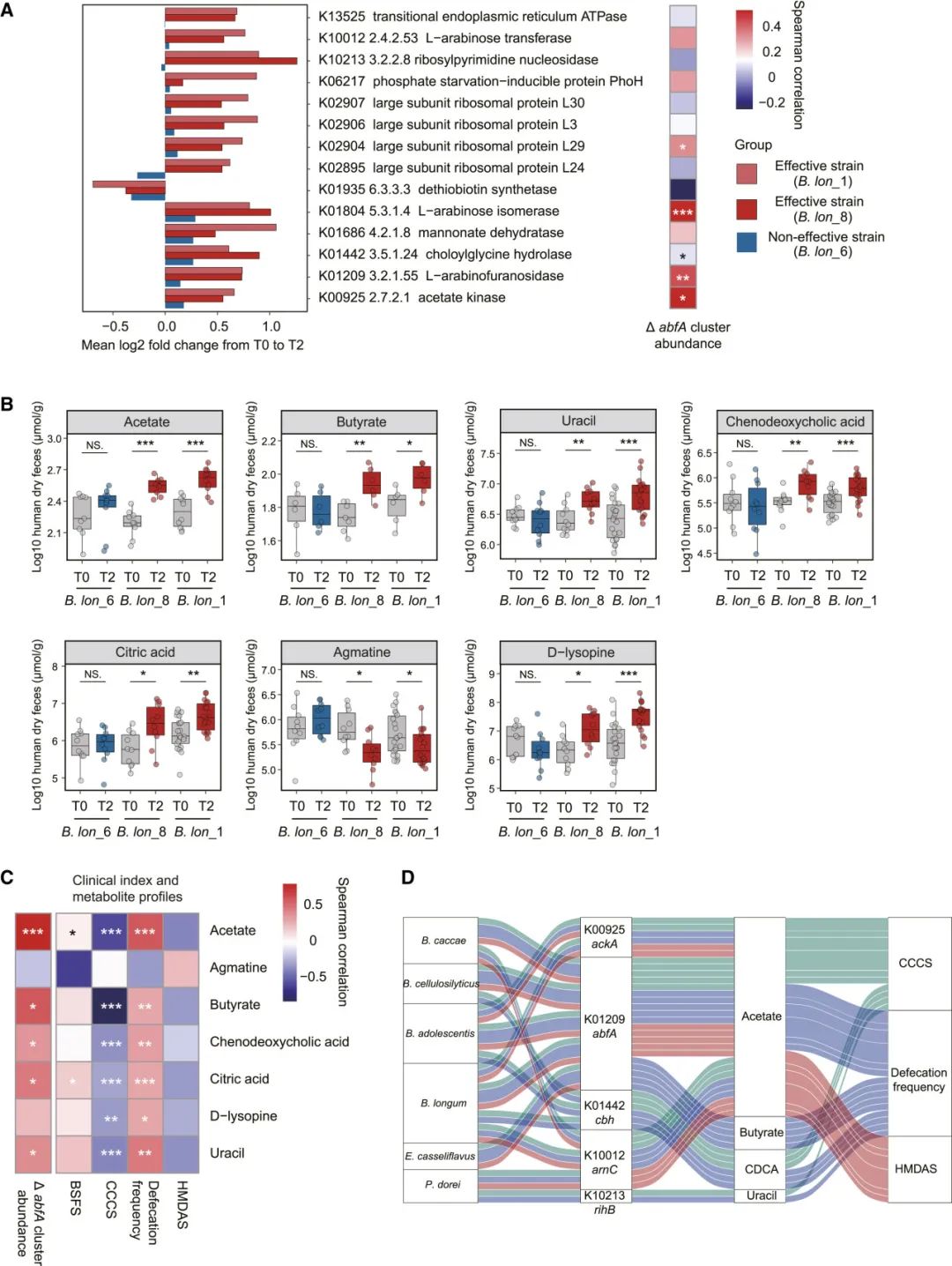
图6 | 长双歧杆菌(abfA+)通过调节本土肠道微生物代谢途径和增强有益代谢物的产生来缓解便秘(FC)
(A) 显示T0和T2之间肠道微生物KOs丰度变化的条形图。相应的热图展示了KOs的时间变化与肠道微生物abfA簇丰度之间的Spearman相关性。
(B) 益生菌消费后28天内基线与代谢物显著差异的肠道微生物代谢物(B. lon_6、B. lon_8和B. lon_1组的代谢物谱分别为n = 10、10和20)。∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001 通过Wilcoxon符号秩检验确定。
(C) 肠道代谢物、临床指标和粪便abfA簇丰度之间的Spearman相关性。
(D) Sankey图总结了微生物菌株、微生物功能基因、粪便代谢物和临床指标之间的关联,基于Spearman相关性 > 0.3。
为了进一步探讨abfA簇介导的粪便代谢物变化,我们首先使用气相色谱-质谱联用(GC-MS)测定了粪便短链脂肪酸(SCFAs)的浓度。结果显示,经过4周B. lon_1和B. lon_8治疗后,两种主要由微生物来源的SCFAs(丁酸盐和醋酸盐)显著增加(Wilcoxon符号秩检验,p < 0.05;图6B;数据S5),而在B. lon_6组中,SCFAs浓度未发生变化。接着,通过非靶向代谢组学(液相色谱-质谱联用[LC-MS]),我们识别了其他与益生菌补充相关的粪便代谢变化。尽管尿嘧啶、陈醋酸、柠檬酸和D-赖氨酸在粪便样本中富集,但在接受B. lon_8和B. lon_1补充治疗的患者中,阿根廷酸浓度降低(Wilcoxon符号秩检验,dFDR调整后的p < 0.1;图6B;数据S5)。然后,我们尝试将宿主内abfA簇丰度的变化与粪便代谢物相关联,揭示肠道内生态与代谢变化的潜在一致性。结果表明,abfA簇丰度的时间变化与粪便中醋酸、丁酸、陈醋酸、柠檬酸和尿嘧啶的浓度变化呈正相关(Spearman秩相关,r > 0.3,FDR调整后的p < 0.05;图6C)。每个宿主肠道中醋酸、丁酸、陈醋酸、柠檬酸和尿嘧啶的浓度升高与排便频率增加(Spearman秩相关,r > 0.3,FDR调整后的p < 0.05;图6C)和CCCS降低(Spearman秩相关,r > 0.3,FDR调整后的p < 0.05;图6C)相关。值得注意的是,在基线时,我们还观察到这些肠道微生物代谢物与宿主间FC症状的差异(即排便频率和CCCS)之间有显著相关性(图S7E)。
最后,我们整合了来自小鼠模型和人类研究的多组学信息,以推测abfA驱动的肠道微生物、代谢物和宿主表型之间的相互作用(图6D)。
FMT实验验证了abfA簇在防治FC中的保护作用
来自健康人类供体的粪便样本(n = 6),包括高丰度abfA簇组(平均相对丰度 = 0.926%,n = 3)和低丰度abfA簇组(平均相对丰度 = 0.031%,n = 3),被移植到两组具有FC的小鼠(n = 6/组)中(图7A)。这两组供体粪便样本在abfA簇丰度上平均存在29.86倍的差异。因此,移植了abfA簇富集粪便的GF小鼠中的abfA簇丰度(平均相对丰度 = 0.89%)明显高于移植了abfA簇缺乏粪便的GF小鼠(平均相对丰度 = 0.028%)(图7B)。如预期,移植了abfA簇丰富粪便的GF小鼠表现出明显的FC缓解,整体肠道通行时间减少了50分钟,粪便输出增加了每小时两块(图7C和7D)。接受abfA簇缺乏粪便菌群移植的GF小鼠没有显著的表型变化(图7C和7D)。接下来,我们分析了GF受体的粪便代谢组,并使用偏最小二乘判别分析(PLS-DA)观察到组间明显的差异(图7E;数据S5)。有趣的是,两组小鼠之间差异丰富的代谢物与人类样本中的差异一致。例如,醋酸、丁酸、陈毒胆酸、D-赖氨酸和尿嘧啶在FMT有效组小鼠中富集(Wilcoxon符号秩检验,dFDR调整的p < 0.1;图7F–7J;数据S5)。这些数据进一步证明,来自本地肠道菌群或外源性益生菌的abfA簇在FC防治中发挥关键的保护作用。
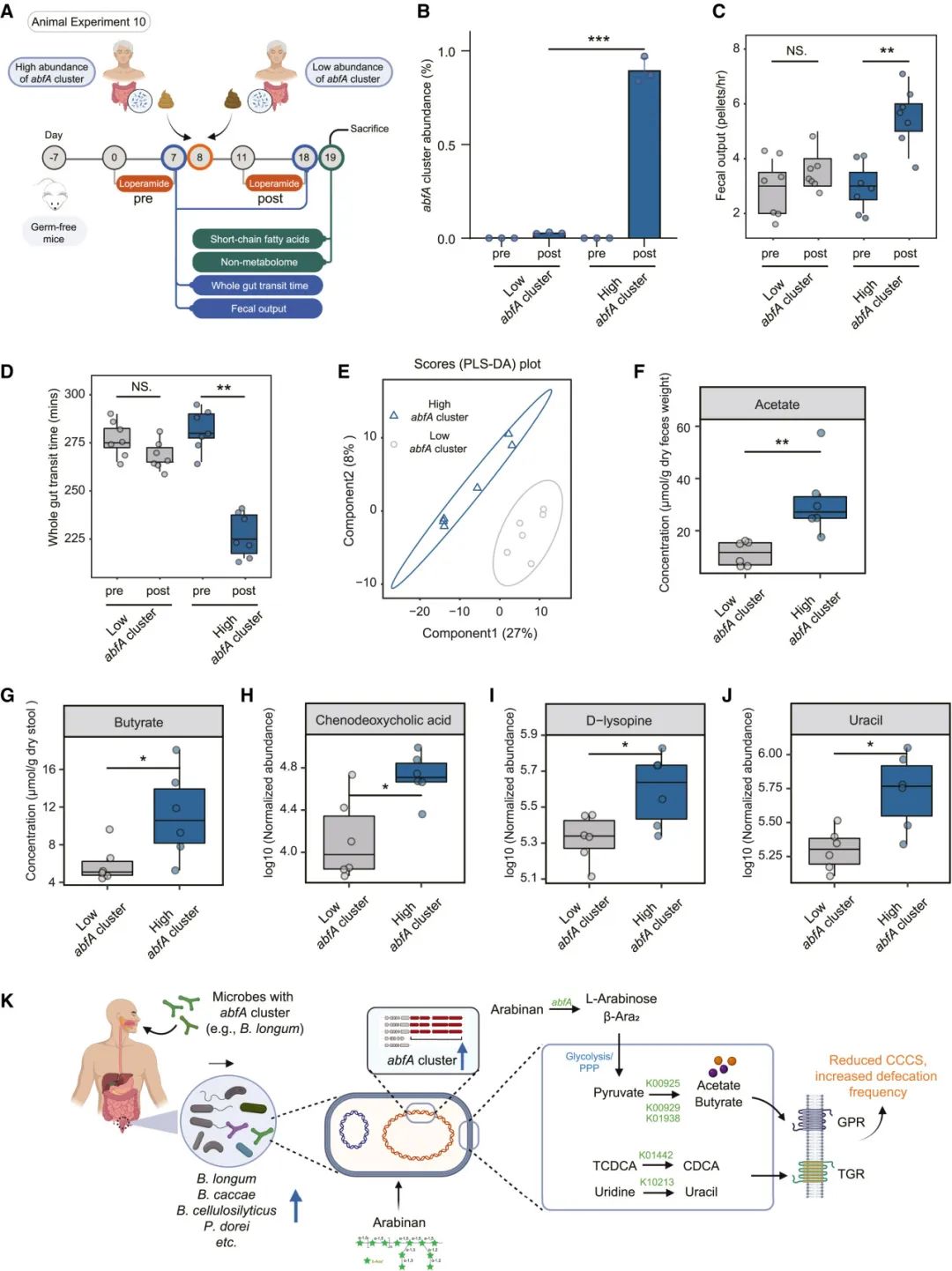
图7 | 移植高丰度abfA簇的粪便菌群缓解了由洛哌丁胺诱导的GF小鼠FC
(A) 实验设计。
(B) 移植后的GF小鼠粪便中abfA簇的丰度。
(C 和 D) GF小鼠移植高丰度或低丰度abfA簇粪便后的 (C) 粪便排出量和 (D) 整个肠道通行时间的变化。
(E) 基于GF小鼠粪便样本的代谢组数据(n = 6–7/组)的部分最小二乘判别分析(PLS-DA)。
(F 和 G) 各组在第19天时粪便中的 (F) 醋酸盐和 (G) 丁酸盐浓度。
(H–J) 各组在第19天时粪便代谢物的归一化丰度。
(K) 总结图,显示abfA簇在缓解FC中的功能作用。
∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,使用Wilcoxon秩和检验(C 和 D)或t检验(F–J)确定。(A) 和 (K) 使用BioRender.com创建。
讨论Discussion
决定益生菌独特代谢能力的遗传因素应作为筛选益生菌或推断其治疗疾病效果的首要考虑因素。益生菌菌株常被经验性地用于治疗功能性便秘(FC),因为之前的宏基因组研究中观察到在FC患者中双歧杆菌、乳酸乳杆菌、植物乳杆菌等的丰度降低。我们在这里提出,单一基因簇可以决定或划分益生菌菌株(例如长双歧杆菌)的关键功能特征,这些菌株在分类学上相同或基因上非常接近。以往的几项研究也表明,单一基因或基因簇可以影响益生菌菌株在体内的细胞表面组成或代谢物谱,并与特定的生理功能(如细菌附着能力、酸耐受性、胆盐耐受性等)相关联。例如,双歧杆菌BAtg基因簇编码的ATP结合盒型碳水化合物转运蛋白,通过在远端结肠代谢果糖生成醋酸,从而保护小鼠免受由E. coli O157:H7引起的死亡。这激发了我们识别决定益生菌双歧杆菌提高肠道运动性的关键遗传因子。在我们之前的研究中,“有效”益生菌菌株通常从有限的候选菌株池中识别出来。这里,通过建立一个包含配对基因组数据的全面菌株库,成功实现了大规模的遗传筛选。接下来,我们前所未有地建立了遗传变异(abfA簇)与益生菌长双歧杆菌在多个模型生物(包括小鼠和人类)中的关键功能差异之间的因果联系,并提供了机制和生态学见解,说明单一基因簇如何通过阿拉伯糖代谢影响宿主的肠道运动性。
abfA 基因簇编码了多个相互关联的α-L-阿拉伯呋喃糖苷酶,这是一种糖苷水解酶,负责分解阿拉伯糖中的糖键。阿拉伯糖是果胶多糖的常见组成部分,也被称为对人类不可消化的纤维或正常肠道菌群难以利用的营养来源。众所周知,由于“优先效应”,外源微生物在肠道生态系统中建立定殖时面临挑战,需要通过创建独特的代谢生态位来与其他菌群竞争。abfA 基因簇赋予长双歧杆菌(B. longum)独特的阿拉伯糖利用能力,有助于其适应和定殖于宿主肠道。
基于稳定定殖,益生菌可以进一步功能性调节便秘患者(FC)肠道菌群。在小鼠和人类的研究中,益生菌定殖促进了具有类似植物多糖代谢能力的本地菌群的生长。有趣的是,这些肠道微生物衍生的有益代谢产物包括乙酸、丁酸、鹅脱氧胆酸和尿嘧啶。乙酸和丁酸是两种众所周知的短链脂肪酸(SCFA),是双歧杆菌和拟杆菌碳水化合物代谢的终产物,它们经常通过与G蛋白偶联受体41(GPR41)和G蛋白偶联受体43(GPR43)相互作用或直接作用于结肠平滑肌来改善肠道蠕动。
这些研究结果支持了我们认为携带abfA 的长双歧杆菌 通过建立特定代谢生态位并进一步增强SCFA生产途径来缓解便秘的观点。此外,双歧杆菌和拟杆菌的丰度还与肠道中的胆汁酸水平(如鹅脱氧胆酸)相关。在我们的研究中,这两类菌群的丰度增加与胆酰甘氨酸水解酶(K01442)水平升高略有相关,该酶主要促进肠道菌群中胆汁酸代谢途径的富集。
对于患有便秘型肠易激综合征(IBS-C)的患者,据报道,使用鹅脱氧胆酸治疗可以通过作用于肠上皮细胞上的膜结合型G蛋白偶联胆汁酸受体(如TGR5)来增加排便频率、降低粪便硬度并改善排便通畅度。
此外,携带abfA 基因簇的长双歧杆菌 定殖后,微生物尿嘧啶代谢也得到增强,这与含有rihB(K10213,参与尿嘧啶代谢的基因)的P. dore 丰度增加有关。类似地,以往的研究也报道了微生物诱导的尿嘧啶水平增加与IBS-C疾病活动减轻之间的关联。
综合来看,在结肠近端,葡萄糖供应通常足以支持微生物生产SCFA、胆汁酸和尿嘧啶;而在结肠远端,葡萄糖供应往往耗尽。在这种情况下,只有携带abfA 基因簇的微生物能够稳定增殖并生产上述有益代谢产物。
微生物生物标志物的发现是人类肠道微生物组研究中的一个重要目标,有助于在临床实践中开发快速、非侵入性的诊断或预后方法。我们证明了肠道菌群中abfA 基因簇的丰度能够高度预测功能性便秘(FC)症状的严重程度,并有可能成为评估抗便秘治疗效果的方法。类似地,Wirbel 等人报道,肠道微生物基因baiF 的拷贝数能够显著区分结直肠癌(CRC)患者和健康对照组(p = 0.001),其受试者工作特征曲线(AUROC)下面积为0.77,这表明使用肠道菌群中的单基因标志物预测胃肠道健康状况是可行的。
综上所述,本研究鉴定并系统性地表征了一个关键的遗传因子,即负责阿拉伯糖利用的abfA 基因簇,解决了益生菌领域中的一个关键挑战:益生菌治疗效果的广泛但未知的菌株特异性。从技术角度来看,我们的概念验证研究还建立了普遍适用的原则,用于合理开发具有持久治疗效果的可定殖功能性益生菌,并在多种模型生物中验证其有效性。
具体而言,基因组中携带abfA 基因簇的益生菌通过创造代谢生态位,能够稳定生产对功能性便秘(FC)有益的代谢产物,从而相较于其他肠道菌群获得适应性优势。此外,abfA 在肠道菌群中十分普遍,可开发为一种简单而有效的FC及其他胃肠道相关疾病的生物标志物。另一方面,我们还强调了检验并恢复人类肠道菌群在膳食纤维(如abfA 基因簇)消化方面的潜在遗传缺陷的重要性,这对解决许多尚无法根本解决的慢性疾病或肠道微生物失衡问题具有重要意义。
实验模型与受试者详情
雄性 SPF BALB/c 小鼠(8周龄,26–28克,来自中国上海斯莱克实验动物有限公司)被用于动物实验。小鼠在标准环境条件下适应了一周(温度:25±2°C;湿度:55%±5%;12小时光暗循环)。实验期间,小鼠可自由获得标准商业鼠粮和无菌水。动物实验方案已获江南大学伦理委员会或江苏省寄生虫病研究所伦理委员会批准(批准编号:JN.No20180615b0950901,JN.No20190315b0940423,JN.No20190315b0940423,JN.No20200710b0700820,JN.No20210630b0400820,lACUC-JIPD-2023101),并按照《欧洲共同体指令2010/63/EU》制定的指南执行。
动物
动物实验 1:长双歧杆菌 菌株效果评估
为了评估 长双歧杆菌 菌株对洛哌丁胺诱导功能性便秘(FC)的影响,80只小鼠被随机分为八组。所有动物在首次实验前禁食过夜(约12小时),但饮水未受限制。随后,小鼠通过灌胃方式每天一次连续14天给予洛哌丁胺(10 mg/kg)以诱导便秘。在洛哌丁胺给药后1小时,通过相同方法将五种 长双歧杆菌 菌株(B. lon_70、B. lon_4、B. lon_8、B. lon_6 和 B. lon_39)以足够剂量(1.0 × 10⁹ CFU/天)灌胃给药。对照组则给予灭菌PBS(0.01 M,pH 7.4),阳性药物对照组给予单剂量酚酞(7 mg/kg)。此外,正常组的小鼠仅给予灭菌PBS(0.01 M,pH 7.4),不接受洛哌丁胺处理。
动物实验 2:阿拉伯聚糖降解基因簇在 长双歧杆菌 菌株中的关键作用
为了评估携带两个阿拉伯聚糖降解基因簇的特定 长双歧杆菌 菌株对洛哌丁胺诱导小鼠功能性便秘(FC)的影响,SPF BALB/c 小鼠(每组 6∼8 只独立生物个体)在洛哌丁胺给药后 1 小时接受以下处理:分别给予 8 种 长双歧杆菌 菌株(B. lon_14 (abfA+, hypBA+)、B. lon_8 (abfA+, hypBA+)、B. lon_56 (abfA+, hypBA-)、B. lon_1 (abfA+, hypBA-)、B. lon_28 (abfA-, hypBA+)、B. lon_58 (abfA-, hypBA+)、B. lon_6 (abfA-, hypBA-)、B. lon_30 (abfA-, hypBA-)、B. lon_32 (abfA+, hypBA+)、B. lon_63 (abfA+, hypBA-) 和 B. lon_64 (abfA-, hypBA-))或灭菌 PBS(0.01 M,pH 7.4)作为对照组。正常组未接受洛哌丁胺处理,仅给予灭菌 PBS(0.01 M,pH 7.4)。
动物实验 3:阿拉伯聚糖与 长双歧杆菌 菌株中 abfA 基因簇在缓解 FC 方面的协同作用
为了进一步评估特定 长双歧杆菌 (abfA+) 菌株对洛哌丁胺诱导小鼠 FC 的影响,SPF BALB/c 小鼠(每组 5∼8 只独立生物个体)分别喂养标准饮食或无阿拉伯聚糖饮食,并接受以下处理:洛哌丁胺联合 B. lon_8、B. lon_6、阿拉伯聚糖+B. lon_8、阿拉伯聚糖+B. lon_6 或仅阿拉伯聚糖。
标准饮食(产品编号:XT93G;4.0 kcal/g)和无阿拉伯聚糖饮食(产品编号:XT19008;4.21 kcal/g;将标准饮食中的玉米淀粉和纤维素-BW200替换为葡萄糖和麦芽糊精-10)由江苏协同药业生物工程有限公司提供。标准饮食包含 7% 脂肪、20.30% 蛋白质和 63.95% 碳水化合物,而无阿拉伯聚糖饮食包含 7.37% 脂肪、21.37% 蛋白质和 67.32% 碳水化合物。阿拉伯聚糖(Megazyme;CAS 号:11078-27-6;来源:甜菜浆)由上海源叶生物科技有限公司提供。
动物实验 4:长双歧杆菌菌株不阻碍蔗糖消化
为了进一步探究长双歧杆菌是否通过阻碍蔗糖消化来缓解功能性便秘(FC),SPF BALB/c 小鼠被给予洛哌丁胺处理,并分别接受盐水、阿拉伯聚糖、B. lon_8 或阿拉伯聚糖+B. lon_8 处理(每组 n=6 个独立生物个体)为期 14 天。
动物实验 5:携带 abfA 基因簇的 长双歧杆菌、FC 症状变化、果糖消耗、阿拉伯糖产生及乙酸生成之间的因果关系
为了验证携带 abfA 基因簇的 长双歧杆菌 的施用与 FC 症状变化、果糖消耗、阿拉伯糖产生及乙酸生成之间的因果关系,6 只 SPF BALB/c 小鼠在 7 天内给予盐水处理,接着 7 天给予洛哌丁胺处理,如此循环 3 次,共持续 42 天。在整个实验中,每个周期包括 7 天的盐水处理后接 7 天的洛哌丁胺处理。在第 7 至 14 天及第 35 至 42 天洛哌丁胺处理阶段,额外给予 B. lon_8 以控制 FC。
动物实验 6:携带 abfA 基因簇的 长双歧杆菌 菌株不会引发健康小鼠腹泻
为了探讨携带 abfA 基因簇的 长双歧杆菌 菌株在未接受洛哌丁胺处理的健康小鼠中的 FC 指标变化,并确认这些菌株是否会引发腹泻,SPF BALB/c 小鼠(每组 n=5 个独立生物个体)分别给予 15 种不同的 长双歧杆菌 菌株(B. lon_14、B. lon_8、B. lon_4、B. lon_70、B. lon_32、B. lon_63、B. lon_56、B. lon_1、B. lon_3、B. lon_28、B. lon_58、B. lon_6、B. lon_30、B. lon_39 和 B. lon_64)。
动物实验 7:基因敲除实验显示 abfA 基因簇在 长双歧杆菌 缓解 FC 中的功能作用
为验证 abfA 基因簇在 长双歧杆菌 缓解功能性便秘(FC)中的重要作用,SPF BALB/c 小鼠(每组 n=6 个独立生物个体)被给予洛哌丁胺处理,并分别接受 B. lon_8(原始菌株)或 B. lon_8ΔabfA(基因敲除菌株)处理,为期 14 天。
动物实验 8:abfA 基因簇在其他肠道本地菌中的功能作用
为了评估含有或不含 abfA 基因簇的更多肠道微生物菌株(如 长双歧杆菌、B. breve、P. pentosaceus 和 B. nordii)对洛哌丁胺诱导的 FC 的疗效,SPF BALB/c 小鼠(每组 n=6∼8 个独立生物个体)被给予洛哌丁胺处理,并分别接受以下菌株处理:B. lon_3 (abfA-)、B. lon_1 (abfA+)、B. bre_41 (abfA-)、B. bre_27 (abfA+)、P. pen_35 (abfA-)、P. pen_16 (abfA+)、B. nor_19 (abfA-) 和 B. nor_12 (abfA+),或以灭菌 PBS(0.01 M,pH 7.4)作为对照组。此外,不给予洛哌丁胺处理的小鼠也用灭菌 PBS(0.01 M,pH 7.4)作为正常组。
动物实验 9:有效与无效 长双歧杆菌 菌株在小鼠粪便及结肠内容物中的丰度比较
为了比较有效与无效 长双歧杆菌 菌株在小鼠粪便及结肠内容物中的丰度,SPF BALB/c 小鼠(每组 n=5∼6 个独立生物个体)被给予洛哌丁胺处理,并接受有效菌株(B. lon_8 和 B. lon_1)或无效菌株(B. lon_6 和 B. lon_3)处理,为期 14 天。在第 7 天和第 14 天评估 长双歧杆菌 菌株对小鼠 FC 指标的影响,同时测量第 0 天、第 7 天和第 14 天小鼠粪便中以及第 7 天和第 14 天结肠内容物中的 长双歧杆菌 菌株丰度。
动物实验 10:高丰度或低丰度 abfA 基因簇携带的人类粪便菌群移植到无菌(GF)小鼠
使用灭菌的磷酸盐缓冲液(PBS),在厌氧环境下放置一夜以去除氧气。将 10%(w/v)新鲜解冻的人类粪便悬浮于预先还原的 PBS 中,并在厌氧条件下旋转 5 分钟。随后以 1000 g 离心 3 分钟,再用 100 μm 的灭菌滤器过滤,将悬浮的细菌与纤维材料分离。在厌氧条件下,将所得的粪便混合物(300 μL)置于一个冷冻管中。8-9 周龄的雄性或雌性 C3H GF 小鼠被随机分为两组(每组 n=6∼8 只),然后通过口服灌胃给予洛哌丁胺,剂量为 10 mg/kg(最大 250 μL)。为避免雄性和雌性小鼠共同饲养,每组中雄性或雌性小鼠(n=3∼4 只)分别放置在独立通风的笼子中,并自由获得灭菌水和鼠类饲料。每 7 天将小鼠放入清洁的笼子中,笼内配有灭菌床垫、棚子、巢穴、水和食物。实验结束后,小鼠通过二氧化碳安乐死并颈部脱位处死。
人类志愿者
共有120名老年参与者从江南大学附属医院招募,其中33名参与者被排除。参与者年龄在65至105岁之间,且在研究前已提交书面知情同意书,受邀接受资格评估程序。符合以下标准的受试者被纳入:i) 满足罗马 IV 便秘诊断标准,包括自述每周排便次数少于3次且根据布里斯托尔大便形态量表自述大便一致性为1-4型;ii) 体质指数(BMI)在18.5-29.9 kg/m²之间。研究中还招募了10名健康的、与便秘患者在年龄和BMI上匹配的受试者。
排除标准包括:i) 有慢性胃肠或神经系统疾病史或其他可能影响肠道蠕动的疾病;ii) 曾接受过重大腹部手术(特别是胃肠手术);iii) 在过去28天内使用与消化道疾病相关的药物(抗痉挛药、抗腹泻药或泻药);iv) 中度或重度肛门直肠问题;v) 对实验益生菌产品成分过敏或乳糖不耐受;vi) 在过去28天内使用过抗生素;vii) 有出血风险或服用增加出血风险的药物。
最终,87名被诊断为便秘的老年人完成了便秘的初始医院管理标准化方案后,参与了本次试验。这些参与者随后被随机分配到四个治疗组之一,并接受了每天一次的治疗,分别为安慰剂(每天2克麦芽糊精;n=21)、B. lon_6(每天1×10⁹ CFU;n=22)、B. lon_8(每天1×10⁹ CFU;n=22)或B. lon_1(每天1×10⁹ CFU;n=22)补充剂,持续28天(图4A)。老年参与者在第一次就诊时需要在适当的协助下填写有关基本健康信息、饮食结构和生活状况的问卷。在每次研究访问时,参与者还填写了评估整个研究期间便秘相关功能性表型变化的问卷,包括使用布里斯托尔大便形态量表(BSFS)评估大便一致性、通过每日大便日志评估排便频率、克利夫兰临床便秘评分(CCCS)、便秘症状患者评估(PAC-SYM)、7天排便日志和便秘生活质量评估(PAC-QoL)。在基线时,研究参与者的性别、年龄、种族、民族、BMI、克利夫兰临床便秘评分(CCCS)和排便频率等参数在四个组之间无显著差异。然而,研究参与者的社会经济状况没有被记录。有关研究对象的性别、种族、民族和社会经济状况的信息见数据S2。
在整个研究过程中,每位受试者都使用家庭采样工具包收集了大便样本。样本管立即保存在冷冻凝胶包中,并在24小时内送回江南大学附属医院胃肠科,样本立即存储在−80°C。健康对照组(n=10)的每位受试者仅提供了一个大便样本,并采用相同的采样协议。
临床试验获得江南大学附属医院医学伦理委员会批准(IEC2020061204和IEC2020092903)。人类大便样本来自已提供知情同意的个体。完整的临床试验注册信息已存档于中国临床试验注册中心(ChiCTR2000034415和ChiCTR2100041925,http://www.chictr.org.cn/)。
方法Methods
长双歧杆菌分离与基因组测序
本研究从中国17个省份或直辖市收集了354份粪便样本用于分离长双歧杆菌菌株。从中国粪便样本中分离出了185株长双歧杆菌菌株,并对其全基因组进行了测序。菌株在mMRS平板(补充0.05% [w/v] L-半胱氨酸和50 mg/mL mupirocin)中培养,在Whitley DG250厌氧工作站(Don Whitley Scientific Limited,Shipley,UK)中孵育48小时,温度为37°C。然后使用DNA提取试剂盒(OMEGA,Biotech,Doraville,GA,USA)从细菌培养物中提取基因组DNA。基因组测序通过Illumina HiSeq平台进行,生成350-bp成对末端读取。使用SOAPdenovo v2.0.455对成对末端读取进行de novo组装,生成高质量的序列。GapCloser用于填补每个骨架内部的空隙并纠正单碱基错误。
长双歧杆菌菌株的系统发育分析
使用Prokka v1.14.6对185个长双歧杆菌基因组进行了重新注释,并通过Roary v3.13.060进行了泛基因组分析,以识别这些长双歧杆菌的核心基因。下载了长双歧杆菌 JCM 1217(NC_015067.1)的基因组作为参考基因组。使用Glimmer v3.02.58预测每个测序基因组的基因CDS。为了获得功能注释,预测的CDS的氨基酸序列通过BLASTP与NCBI nr和COG数据库进行比对,标准为e值<1e-5,序列相似度>40%,基因长度覆盖率>50%。使用默认参数(氨基酸水平上相似度50%)的OrthoMCL v2.0.9来识别有效和无效菌株基因组中的同源基因。通过卡方检验(p<0.05)识别有效和无效长双歧杆菌菌株基因组中显著不同的功能基因或基因簇,并进一步分析这些功能基因或基因簇在不同菌株(有效或无效菌株)中的分布,以确定在有效长双歧杆菌菌株中特有的基因或基因簇。使用R包“gggenes”绘制了针对有效益生菌菌株特有的abfA和hypBA基因簇的基因箭头图。通过FastANI v1.32.57计算新测序基因组之间的成对ANI值。
分离菌株的生长检测
将菌株接种在补充1.0% D-葡萄糖的糖限制基础培养基中,并在37°C的厌氧条件下培养,直到600 nm处的光密度(OD600)达到1.0。然后将5%体积的种子培养液加入含2.0%阿拉伯糖或2.0% D-葡萄糖的基础培养基中。将这些混合物加入96孔板,并用Microseal B板封膜(Bio-Rad,美国)封闭。将培养板保持在37°C,并每2小时使用Powerscan HT plate读数仪(大日本住友制药,日本)监测OD600。
基因簇的转录活性
B. lon_8菌株在37°C的厌氧条件下在含2.0%阿拉伯糖或2.0% D-葡萄糖的糖限制基础培养基中培养。通过离心(8000 g,10分钟)在无酶管中收集1 mL的培养液,然后与200 μL溶菌酶溶液混合。将混合物在37°C下孵育30分钟以破坏细胞壁。无细胞壁的细胞通过离心(12000 g,5分钟,4°C)收集。使用TRIzol Plus RNA纯化试剂盒根据制造商的协议提取每个菌株的总RNA(Invitrogen,美国加利福尼亚州)。使用NanoDrop分光光度计(Thermo,上海,中国)测量RNA浓度。使用HiFiScript gDNA去除cDNA合成试剂盒(CoWin Biosciences,江苏无锡,中国)在20 μL反应体系中合成cDNA。按照TransStart Tip Green qPCR SuperMix试剂盒(Tiangen Biotech Co.,Ltd.,北京,中国)的说明进行qRT-PCR实验。使用Primer5软件(美国加利福尼亚州)设计特定引物,并由Sangon Biotechnology Laboratory(上海,中国)合成。16S rDNA被用作参考基因。每个实验均进行三次重复。
时间序列共培养实验
B. adolescentis 菌株442(B. ado_442)、E. casseliflavus 菌株M598(E. cas_M598)、B. cellulosilyticus 菌株BHI5(B. cel_BHI5)、P. dorei 菌株K4(P. dor_K4)和B. cacca 菌株MIC3(B. cac_MIC3)由江南大学食品微生物文化收藏室(中国无锡)提供。在共培养之前,每个拟杆菌属(Bacteroides spp.)和肠球菌属(Enterococcus spp.)菌株在改良的脑心浸液培养基(BHI)中培养,双歧杆菌属(Bifidobacterium spp.)菌株在改良的德曼·罗戈萨·夏普(mMRS)培养基中培养,并在PBS中洗涤后,用于接种含0.5%阿拉伯糖的GMM培养基。在中后期对数生长期收获的细菌菌株被重新悬浮在调整到约105 CFU/mL的GMM培养基中。共培养在三重复条件下进行,并在0小时、3小时、6小时、9小时、12小时、18小时和24小时定期取样,分析碳水化合物降解、微生物生长(OD600)和短链脂肪酸(SCFAs)。从1 mL的样品中提取DNA,并通过特异性引物进行qPCR分析。qPCR反应体系(总量20 μL)包括TransStart Tip Green qPCR SuperMix试剂盒(Tiangen Biotech Co., Ltd.,北京,中国)、2 μL模板DNA和0.2 μM的每个引物。扩增条件包括初始变性:94°C,10秒;然后进行40个循环:94°C,20秒;55°C,20秒;72°C,50秒;最后进行熔解曲线程序。扩增使用CFX Connect实时PCR检测系统(Bio-Rad,美国)进行。标准曲线通过使用每个菌株提取的DNA的十倍稀释系列进行计算,以进行细胞数的绝对定量。
引物信息:
长双歧杆菌 (F: 5′- TTCCAGTTGATCGCATGGTCTTCTA-3′, R: 5′- GGCTACCCGTCGAAGCCACG-3′)
B. breve (F: 5′- CCGGATGCTCCATCACACG-3′, R:5′- ACAAAGTGCCTTGCTCCCT-3′)
B. adolescentis (F: 5′- AGCAATCTTCATGGTTGCG-3′, R:5′- ACCGTCTCGGTTTTGCCGGTCCATG-3′)
B. ovatus (F: 5′-GGAATGAGCATAATCCATATATCAAGATGAAACG-3′, R: 5′-TACCTGAAACAATCATCCTTTATTTCTGTAGC-3′)
B. cellulsoylticus (F: 5′-AGCAGGCGGAATTCGATAAG-3′ R:5′-GTGTACAGTGCCAGGCATAA-3′)
B. dorei (F: 5′-ATCATGAGTTCACATGTCCG-3′, R: 5′-CTTCCTCTCAGAACCCCTATCC-3′)
B. nordii (F: 5′-GCGGGGAACAGAATCAGACA-3′, R: 5′-ATTCCACCAAATGTAGGCGGGACGTTTAAT-3′)
E. casseliflavus (F: 5′-AGAAATTCCAAACGAACTTG-3′, R: 5′-CAGCGATCACATTCGCCTTG-3′
细菌突变体构建
通过双重交叉同源重组敲除BLLJ_1852-1853基因区域,参考Sakaguchi等人、Altaib等人和Hirayama等人方法。为了构建基因敲除的质粒,从B. lon_8的染色体上PCR扩增出BLLJ_1852-1853基因上游889 bp和下游870 bp的 flanking 序列。通过重叠PCR将这些DNA片段和1.2 kb的抗链霉素基因(Spr)连接起来。为了获得pKO403-ΔabfA,使用ClonExpress Ultra One Step Cloning Kit(Vazyme,南京,中国)通过同源重组将3.0 kb片段整合到线性化的pKO403中。通过电转化仪2510系统(Eppendorf,上海,中国)将靶向载体(pKO403-ΔabfA)以2.5 kV的电压,2 mm的电转化间隙转导到B. lon_8中。转化子在30°C下,在含有链霉素的mMRS平板上培养72小时。随后,将其转移至含链霉素的mMRS液体培养基中,在30°C下培养24小时。通过离心(3000 g,5分钟)收集培养细胞。这些收集的细胞接着在含链霉素的mMRS平板上涂布,并在42°C下培养24小时,之后在30°C下再培养24小时。最终,在42°C下能生长的菌落转移到含有链霉素和氯霉素的MRS平板上进行复制。这些复制的菌落在37°C下培养24小时。那些表现出抗链霉素和对氯霉素敏感的菌落被选作基因敲除的潜在候选菌株,并通过基因组PCR进一步分析。
粪便宏基因组测序
使用QIAamp DNA Stool Mini Kit(Qiagen,德国希尔登)从粪便样本中提取DNA,进行0.8%琼脂糖凝胶电泳检测,并通过分光光度计测定260/280的OD值。所有DNA样本均使用HiSeq 2500(Illumina,美国加州)进行散装宏基因组测序,采用150 bp的双端读数(正向和反向),由Novogene公司(北京,中国)进行操作。通过FastQC v0.11.961控制测序读数的质量,并随后使用KneadData v0.7.2进行修剪并去除人类序列。
散装宏基因组数据分析
经过质量过滤的宏基因组数据使用MetaPhlAn2 v2.2.0进行分类分析。功能分析使用HUMAnN2 v0.11.2,在UniRef90模式下默认设置进行。因此,我们获得了每个宏基因组的肠道微生物种类、基因家族和代谢途径的相对丰度谱。接下来,使用包括MEGAHIT v1.2.9进行基因组组装、Prodigal v2.6.3进行开放阅读框预测、MetaBAT v2.12.1进行基因组分箱、CheckM v1.1.3进行基因组质量控制的工作流构建了宏基因组组装基因组(MAGs),然后使用GTDB-Tk v1.0.2进行分类鉴定。
Kraken 2分类序列系统经过修改,通过创建定制数据库来追踪益生菌的丰度,并在菌株层面上进行分析。B. lon_8、B. lon_6和B. lon_1的基因组文件以FASTA格式添加到基因组库中,使用--add-to-library开关在分配其分类信息后进行。使用--build开关构建数据库。Kraken 2基于新的定制数据库确定这三种菌株的丰度。
利用LC-MS的代谢组学分析
代谢组学分析采用Liu等人提出的方法进行。提取时,每个小鼠的粪便样本(约50 mg)立即在液氮中迅速冷冻,并与400 µL预冷的超纯水和甲醇混合(1:4,v/v),使用高通量组织匀浆机SCIENTZ-48(SCIENTZ,浙江宁波,中国)在-20°C下匀浆6分钟。然后每个样本振荡30秒,并在低温超声提取下孵育30分钟(5°C,40 kHz);随后存放在-20°C下30分钟。含样品的试管在4°C下以1300 g离心15分钟,获得上清液。质量控制(QC)样本通过将每个粪便样本的重溶液体积均等混合制备,并在每10个粪便样本后分析。QC样本可监控系统的稳定性和可重复性。空白样本(80%甲醇)用于背景扣除和噪音去除。
粪便样本代谢物提取液(上清液)的LC-MS分析使用UltiMateU-3000 UPLC系统(Thermo Fisher Scientific,MA,美国)与高分辨率Q-Exactive质谱仪联用,在全扫描模式下进行(Thermo Fisher Scientific,MA,美国)。采用2.1 × 100 mm反相Waters Acquity UPLC T3色谱柱(Waters,MA,美国)在35°C下进行色谱分离。流动相由0.1%的水合甲酸(A)和0.5 mM乙酸铵-乙腈(B)组成,采用梯度洗脱:0–1.0分钟(5%,B);1.0–10分钟(5%-99%,B);10–12分钟(99%,B);12–15分钟(5%,B)。流速为0.3 mL min−1,进样体积为2 µL。MS系统配备加热电喷雾电离(ESI)源,在正负离子模式下操作,扫描范围分别覆盖151到2000 m/z和70到1050 m/z,参数设置如下:源电压为1.50 kV,毛细管温度为250°C。负离子和正离子化模式的毛细管电压分别设置为2500 V和3500 V。
MS数据通过Compound Discovery 3.1(Thermo Fisher Scientific,MA,美国)使用无靶代谢工作流处理,采用默认参数,包括数据提取、背景特征过滤、峰值识别、解卷积、对齐和积分,以及通过mzCloud、HMDB、KEGG和ChemSpider数据库进行代谢物鉴定。数据处理过滤掉了在QC样本中出现频率低于50%或RSD大于30%的代谢特征,质量公差设为5 ppm。无法匹配上述数据库中质量的m/z比率的代谢特征也被过滤掉。处理后的数据包括峰面积、保留时间(RT)、分子质量(MW)以及已识别或未知化合物的整合。多变量分析使用SIMCA软件(Umetrics,瑞典乌梅阿)进行。代谢途径分析使用MetaboAnalyst 5.0(https://www.metaboanalyst.ca/)进行。
糖类和洛哌丁胺的测定
根据Ruddle等人的方法85进行阿拉伯糖、果糖、蔗糖和洛哌丁胺的测定。取发酵液(约0.5 mL)放入1 mL管中,离心(12000 g,5分钟,4°C)。然后,加入400 μL预冷的甲醇:乙腈(1:1,v/v),振荡30秒,并进行低温超声提取10分钟(5°C,40 kHz)。样品随后在-20°C存放1小时。再次加入400 μL甲醇:乙腈(1:1,v/v)(预冷至-20°C),每个样品再次振荡30秒,进行低温超声提取10分钟(5°C,40 kHz),然后在-20°C存放1小时。含样品的试管进行离心(12000 g,15分钟,4°C)以获得上清液。提取液用甲醇重溶至1 mL,并通过0.2 μm滤膜过滤后进行HPLC-MS/MS分析。
上清液使用UltiMate 3000 UPLC系统(Thermo Fisher Scientific,MA,美国)联用高分辨率Q-Exactive质谱仪(Thermo Fisher Scientific,MA,美国)分析。LC分离使用Waters Acquity UPLC HSS T3柱(2.1 × 100 mm,1.8 μm;Waters, Milford,美国),流动相A(0.1%甲酸水溶液)和流动相B(乙腈)。线性梯度如下:0.1–1.0分钟(99% B);1.0–8.0分钟(99–1% B);8.0–9.0分钟(1% B);9.0–9.1分钟(1–99% B);9.1–12.0分钟(98% B);12.1分钟(2% B);12.1–15分钟(2% B)。流速为0.3 mL/min,进样量为2 μL。样品在自动进样器中保持在4°C,色谱柱在35°C下运行。质谱系统使用加热的电喷雾离子源(ESI),在正负离子模式下工作,扫描范围分别为151至2000 m/z和70至1050 m/z。参数设置如下:1.50 kV源电压和250°C毛细管温度。负离子和正离子化模式的毛细管电压分别设为2500 V和3500 V。
用HPGPC-RID法测定阿拉伯甘露糖
阿拉伯甘露糖的含量通过高效凝胶渗透色谱法联用折光检测器(HPGPC-RID)测定,采用Zhao等人方法的改进版86。HPGPC-RID使用凝胶过滤色谱柱(Ultrahydrogels™ Linea,7.8 mm × 300 mm,Waters,MA,美国),保持在40°C,使用0.1 mol/L Na2CO3溶液进行洗脱,流速为0.5 mL/min。阿拉伯甘露糖(约15000道尔顿)标准品(50.0 mg)溶解在去离子水中(1 mL),通过0.22 μm滤膜过滤后,制备系列标准溶液,并建立了与浓度相关的标准曲线。
用GC-MS法定量短链脂肪酸(SCFAs)
SCFAs的浓度按之前的描述测定42。每个粪便样本0.2克,重新悬浮在饱和NaCl溶液(500 μL)中,加入20 μL硫酸(10%)进行酸化。然后加入800 μL二乙醚提取SCFAs。振荡2分钟后,混合物在14000 g下离心15分钟,再加入0.25 g无水Na2SO4去除内水,并保持在-20°C 30分钟。将上清液转移至新鲜玻璃瓶中,进行GC-MS分析。GC-MS分析使用气相色谱-质谱联用系统(GCMS-QP2010 Ultra系统,岛津公司,日本)。该系统使用Rtx-Wax色谱柱(30 m × 0.25 mm × 0.25 μm,Restek,Evry,法国)。用1 μL样品在分流模式(10:1)下注入。氦气作为载气,前进气口的冲洗流速为2 mL/min,气体通过色谱柱的流速为1 mL/min。初始温度保持在100°C 1分钟,然后以7.5°C/min升至140°C,再以60°C/min升至200°C,最后在200°C保持3分钟。注入、传输线和离子源的温度分别设置为240°C、240°C和220°C。采用单次扫描模式检测分析物(醋酸、丁酸、异戊酸和戊酸:60 m/z;丙酸:57 m/z;异丁酸:43 m/z)。SCFAs的浓度通过计算每种SCFA相对于4-甲基戊酸的响应因子来计算,并以μmol/g干重样品表示。
粪便颗粒水分含量的测定
在一项持续5小时的实验中,每30分钟记录每只小鼠的粪便颗粒数量,以测量小鼠的肠道运动。通过每小时小鼠粪便颗粒的平均数量进行个体间比较。然后,每只小鼠收集一个新鲜的粪便颗粒,放入单独的无菌EP管中。获得湿重后,将每个样本放入冻干机中冻干48小时,获得干重。通过湿重和干重的差值计算每只小鼠的粪便水分含量。
全肠道通过时间的测定
在每次动物实验的终点(例如,第14天),小鼠禁食过夜,并提供饮用水。然后,实验组的小鼠给予了洛哌丁胺(10 mg/kg体重),对照组小鼠给予0.9%的生理盐水。1小时后,所有小鼠口服250 μL的0.5%活性炭溶液,并加入牙膏粉,以减轻活性炭引起的小鼠肠道潜在刺激。随后,将小鼠立即转移到一个干净的空笼中,自由进食和饮水。全肠道通过时间或排便时间被记录为排出第一颗含有活性炭的粪便颗粒所需的时间(即,变色的粪便)。
小肠通过时间的测定
胃肠道运动性使用Verdú等人方法进行测量,并略作修改。87 在禁食12小时后,每只小鼠通过上述方法给予活性炭溶液。首先,为了测量血液来源的胃肠激素,小鼠被麻醉,并用轻度醚麻醉以收集血样。每只小鼠抽取的全血放入试管中静置约2小时,使其凝固。然后通过3000 g离心15分钟分离血清。接下来,打开每只小鼠的腹部,仔细取出整个小肠(从幽门到盲肠),并放置在吸水纸上。测量活性炭移动的距离和小肠的总长度。每只小鼠的小肠通过时间通过将活性炭粉末的移动距离与小肠的总长度相对比计算得出。
定量与统计分析
所有统计分析使用R v 4.2.2进行。对于β多样性分析,计算每对粪便菌群落之间的Bray-Curtis差异,并用于估算环境因素的效应大小,使用Adonis检验。统计显著性通过以下方式确定:Student’s t检验(两组比较,正态分布)和单因素方差分析(比较三组或更多组)。接着使用Wilcoxon秩和检验或Wilcoxon符号秩检验来确定在处理组或时间点中富集或消耗的微生物特征(如微生物属、物种、菌株和功能基因)。热图使用“pheatmap”包(v1.0.12)构建。箱形图使用R包“ggpubr”(v0.2.3)绘制。使用R包“DiscreteFDR”(v1.3.6)识别差异丰度特征,KEGG同源物和代谢组学分析使用dFDR阈值<0.1。通过Spearman等级相关性与FDR调整用于构建分子级特征(微生物分类群、功能基因和代谢物)特征与临床协变量之间的共现网络。使用R包“ggalluvial”(v0.12.5)进一步通过Sankey图可视化它们之间的相互作用。使用R包“gggenes”(v0.5.0)可视化abfA和hypBA基因位点的结构。
作者简介
江南大学食品学院张程程为本文第一作者,江南大学食品学院翟齐啸教授、海南大学张家超教授、香港大学牙医学院黄适助理教授为本文的共同通讯作者。
翟齐啸(通讯作者)

江南大学食品学院副院长、食品科学与资源挖掘全国重点实验室副主任,国家杰出青年基金、优秀青年基金及江苏省优秀青年基金获得者。主要从事益生菌理论与技术、肠道微生物功能挖掘及肠道菌群与功能食品相关研究。近年来以第一或通讯作者在 Cell Host & Microbe、Cell Genomics、PNAS 等期刊发表 SCI 科研论文 150 篇,获中国授权专利 60 项,国际授权专利 8 项。负责主持包括国家自然科学基金联合基金重点、面上、青年,国家十四五重点研发计划课题等科研项目 14 项。《Food Science and Human Wellness》副主编。
张家超(通讯作者)

张家超,博士,教授,博士生导师,现任海南大学食品科学与工程学院执行院长,热带多糖资源利用教育部工程中心副主任,海南省食品营养与功能食品重点实验室副主任、食品学会青委会委员,中国食品科学技术学会益生菌分会理事,获国家优秀青年科学基金资助,入选海南省“南海名家”资助计划和海南自由贸易港高层次人才。主要从事热带益生菌资源开发利用相关研究。近年来承担国家重点研发计划子课题1项、国家自然科学基金3项和省部级项目9项;以第一/通讯作者发表SCI论文70余篇,授权国家发明专利12项;荣获第十届海南省青年科技奖、海南省自然科学二等奖,海南省教学成果二等奖。
黄适(通讯作者)

黄适,近年来作为(共同)第一或通讯作者在Nature Methods,Cell Host Microbe,Microbiome,Genome Biology,ISMEJ,mSystems,Gut Microbes,mBio等国际知名学术期刊发表论文12篇,共计论文超30篇。申请发明专利7项,其中授权国际专利2项、国内专利2项。主持和参加包括国家青年自然基金等多项基金项目。
翻译:荀佳妮,中国农科院基因组所,生物信息学硕士在读
审核:朱志豪,广东医科大学,基因组所联合博士后
终审:刘永鑫,中国农科院基因组所,研究员/博导
排版:杨海飞,青岛农业大学,基因组所联培硕士在读
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 726
726

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









