









- 蛋白质组概述
- 研究意义:
- 蛋白质组是空间和时间上动态变化着的整体,一个基因组对应多个蛋白质组
- 人类与简单生物的巨大差别,来自蛋白质之间相互作用的数量
- 蛋白质定义:一类重要的生物高分子,参与了生物体内几乎所有的生理功能和代谢过程。由20种氨基酸通过肽键(酰胺键)连接形成的长链分子(肽链),在此基础 上,肽链进一步形成二级、三级的空间结构。有的蛋白质还包含辅基成分,如金属铁、 锰等。
- 蛋白质组:一种基因组或一个细胞、组织所表达的全套蛋白质
- 蛋白质组学:围绕一种细胞或一个生物体所表达的全部蛋白质,在大 规模水平上研究蛋白质的特征,包括蛋白质的表达水平, 翻译后的修饰,蛋白与蛋白相互作用等,由此获得蛋白 质水平上的关于疾病发生,细胞代谢等过程的整体而全 面的认识
- 研究意义:
- 研究技术和方案设计
- 定量蛋白质组学
- 同位素标记法
- iTRAQ
- 基本概念:
- iTRAQ(isobaric tags for relative and absolute quantification)技术是由AB SCIEX公司研发的一种多肽体 外标记技术,通过同位素特异性标记多肽的氨基基团,然后进行串联质谱分析进来检测蛋白组中蛋白质的相对含量。
- iTRAQ标记试剂由三部分组成:报告基团、质量平衡基团和 肽反应部分,形成4种或8种相对分子质量均等量的异位标签。
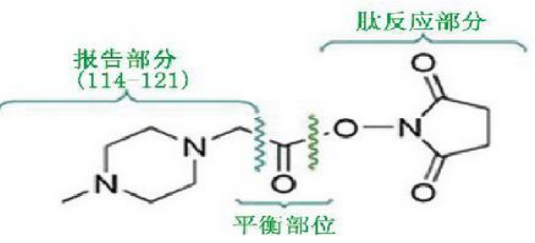
- 1、报告部分:有八种,因此iTRAQ最多可同时标记8个样品
- 2、肽反应部分:能与肽N端及赖氨酸侧链发生共价连接而 标记上肽段,几乎可以标记所有蛋白质。
- 3、平衡部分:保证iTRAQ标记的同一肽段的质荷比相同
- 一级质谱测不同样品的同一蛋白
- 二级质谱测不同样品的含量
- 设计原则
- 生物学重复可分批上机
- 需要比较的差异分组放在一组标记中
- 不同组织部位不要放在一组标记中检测
- 最大利用效率
- 基本概念:
- TMT
- 基本概念:TMT(Tandem Mass Tag)技术是由Thermo公司研发的一种多肽体外标记技术,通过同位素特异性标记多肽 的氨基基团,然后进行串联质谱分析来检测蛋白组中蛋白质的相对含量。
- 与iTRAQ比较:
- 相同点: 原理、实验流程与iTRAQ相同
- 不同点: TMT标记试剂10种,iTRAQ是8种
- iTRAQ更成熟、标记效率更稳定
- iTRAQ标记试剂便宜一些
- iTRAQ
- 非标记法
- label-free
- 通过质谱波峰的高度及面积来进行定量。因其不受样品来源和数量的 限制,因而近年来得以广泛应用。
- 主要步骤包括:蛋白酶解、LC-MS/MS质谱分析、数据分析。
- 实验过程中,每个样品是分别上质谱检测
- 一级MS的信息:峰面积,计算肽段在色谱上的积分
- 二级MS的信息:定性 与标记方法的不同点:
- 无需同位素标记,成本低
- 每个样品分别上机进行检测
- 不受样品数量的限制
- 蛋白量要求少
- DIA
- DDA
- 利用一级全扫描检测肽段母离子,然后按信号强度排列,将前若干位的母离子依次选择碎裂,并扫 描二级碎片离子
- 易造成低丰度肽段信息丢失;母离子选择有一定的随机性,重现性不佳
- DIA
- 将质谱整个全扫描范围分为若干个窗口,高速、循环地对每个窗口中的所有离子进行选择、碎裂、 检测,从而无遗漏、无差异地获得样本中所有离子的全部碎片信息
- 需要先建库,对系统稳定性要求高,谱图解析难度大
- DDA
- PRM (靶向验证)
- 概念:基于质谱的离子检测技术,能够通过特异性检测目标肽段、全扫描检测其所有二级子离子,根 据可靠二级子离子定量实现对目标蛋白质(肽段)进行定量。利用该技术既可实现样品中靶标物质 的相对定量,又可通过合成同位素标准肽段,绘制标准曲线,实现蛋白质的绝对定量。
- 应用:
- 1.用于label free, iTRAQ 筛选得到的差异蛋白质的 验证(目的等同于WB)
- 2.用于生物标志物验证和 评价
- 优势:
- 1.无需抗体
- 2.无物种限制
- 3.一次检测多个定量指标, 通量高
- 4.新颖的验证手段,助力文章影响力
- 注意事项:
- 1.提供蛋白列表50-60个蛋白
- 2.预实验,根据客户提供列表, 筛选可以做PRM的蛋白
- 3.能否进行PRM验证,取决于蛋白特异性肽段数量以及质谱中的 响应情况
- 4.最后能验证20个以内的蛋白
- label-free
- 平台选择
- labelfree
- 样品分组>8、样品数>24、样品不好收集,样本量至少 60ug 实验设计
- 研究蛋白的有无、低丰度的蛋白
- 个体差异比较大、临床样本、涉及不同物种间分析
- iTRAQ
- 样品分组<=8、样品数<=24、样本量至少达到200ug以 实验设计 上
- 关注定量准确 经费要求
- 经费充足、周期长(标记和分级)
- labelfree
- 同位素标记法
- 定性蛋白质组学
- *修饰蛋白组学(不做)
- 定量蛋白质组学
- 项目流程
- 项目流程图
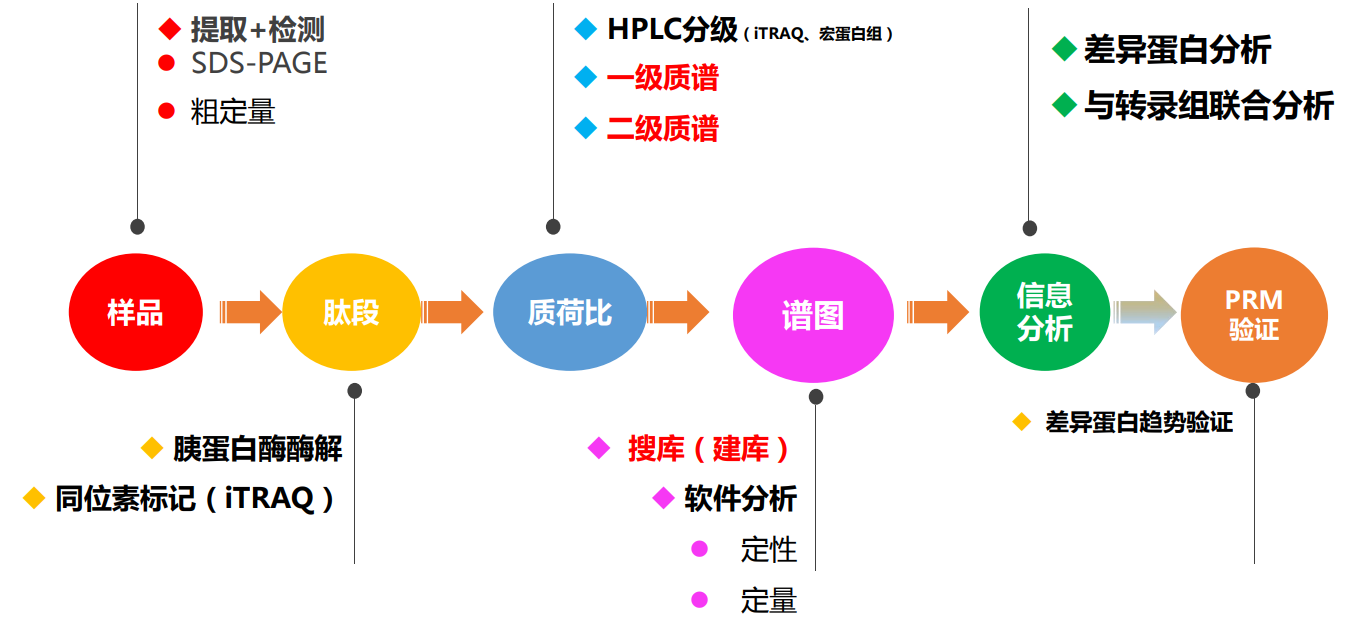
- 质谱研究流程
- Bottom-up研究思路
- 鸟枪法(Shotgun)法
- --数据产生:蛋白->肽段->谱图
- --数据分析:谱图->肽段->蛋白
- 打散:通过酶切,把蛋白质切碎得到肽段混合物;
- 检测:肽段混合物经过色谱分离和离子化后, 经串联质谱碎裂产生实验谱图;
- 搜库
- 首先,我们得选一个蛋白序列数据库,可以是公共数据库,最好是用 转录组数据构建的。
- 其次,把质谱仪得到的谱图输入搜库软件。对于搜库软件来说,碎片 离子的信息越丰富越好。
- 搜库软件通过以下五步来实现谱图的正确匹配:
- 1) 从数据库中选择分子量与输入值相等的肽段;
- 2) 生成理论碎片,并生成理论谱图;
- 3) 将实验谱图与理论谱图进行匹配;
- 4) 对匹配进行打分;
- 5) 将打分进行排序,通过统计学分析,确定最佳的匹配结果并导出。
- 优先级
- 首选,同批样品转录组数据构建的蛋白库(蛋白 鉴定数提升20%-50%)
- 其次,uniprot数据库(或NCBI)选择所研究物种
- 再次,uniprot数据库(或NCBI)选择近源物种
- 质谱研究流程
- 项目流程图
- 转录组+蛋白组联合分析
- 转录组和蛋白质组联合分析技术流程
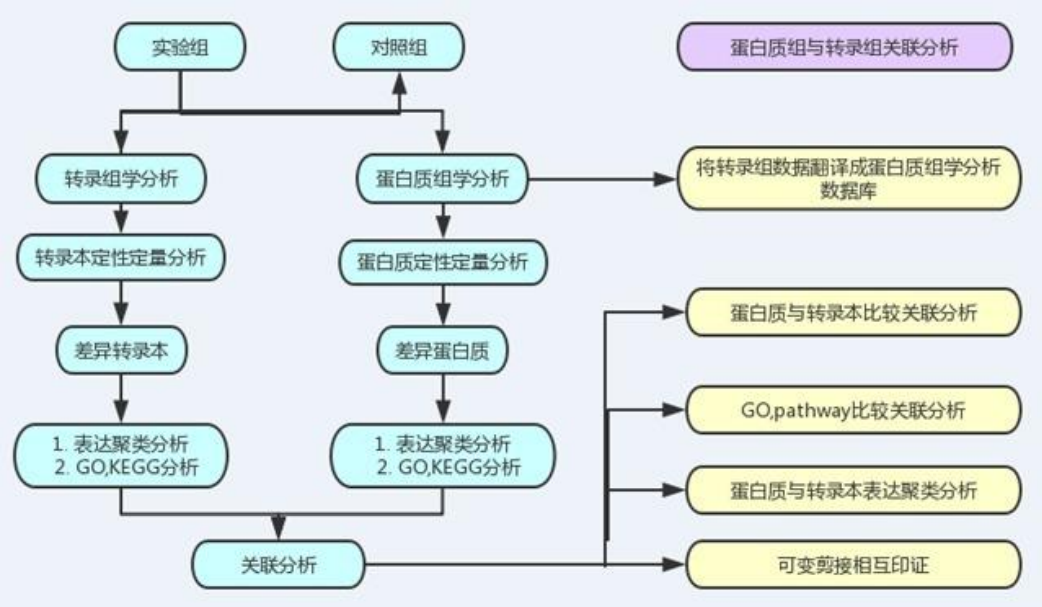
- 1、利用转录组数据构建蛋白搜索数据库
- 利用转录组数据来建立蛋白搜索数据库,这将很大程度上提升肽段及蛋白的鉴定数量。实验表明,基于 转录组数据建立蛋白搜索数据库,蛋白鉴定数量平均可以增加20%~50%, 尤其对于非模式生物来说, 由于相关的研究非常少(公布的基因序列较少),其蛋白质序列数据库质量比较差。
- 2、差异蛋白与差异基因表达水平比较关联分析
- 首先将鉴定到的所有蛋白和与之相对应的基因的转录本进行综合关联比较分析。 ü 在此基础上,根据各自的表达变化的定量信息,对关联上的差异表达蛋白和与之相对应的差 异基因的转录本进行比较分析。
- 3、蛋白组与转录组表达模式聚类分析
- 4、蛋白组和转录组表达趋势GO功能、KEGG的关联比较分析
- 5、其他
- COG分析
- PCA分析
- 蛋白互作网络分析
- 蛋白序列亚细胞定位预测
- 差异蛋白富集弦图
- 定量答案白组文章路线
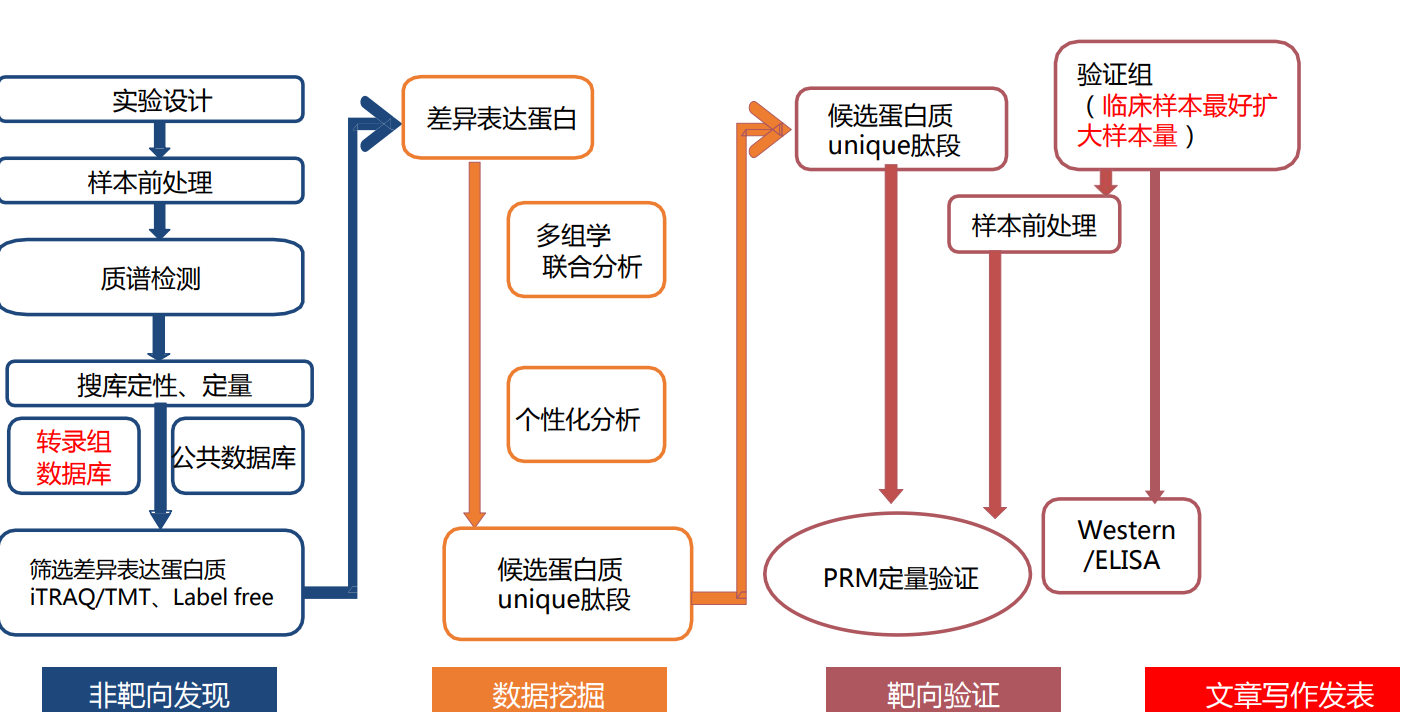
- 转录组和蛋白质组联合分析技术流程
- 应用方向
- 发育研究
- 植物
- 发育研究:比较不同时期、不同组织、器官、发育阶段、不同亚种间比较研究、物质合成、代谢通路的研究、种子发芽率 的研究等。
- 逆境胁迫:了解非生物胁迫的伤害机制、植物对非生物环境的适应机制、生物之间的相互作用机制、植物激素的调节作用 等有重要意义。气候,抗逆,生活习性,外部刺激;
- 动物
- 发育研究:肉质研究、不同品种研究、生长发育研究等
- 医学
- 方案设计
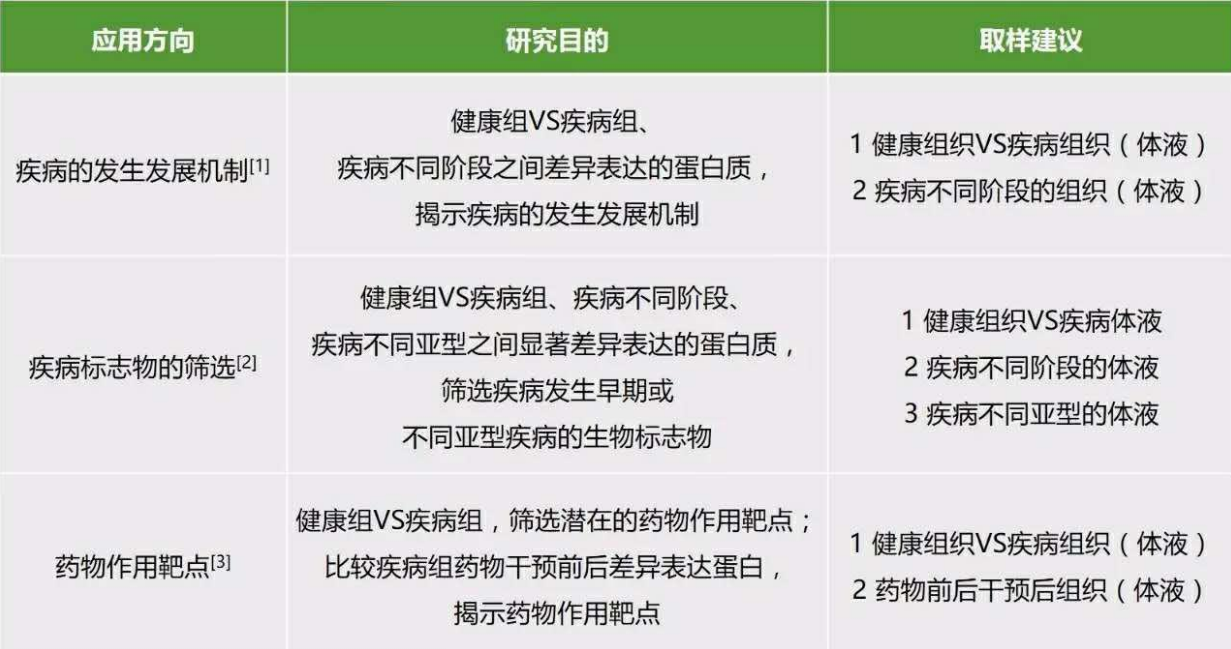
- 方案设计
- 植物
- 发育研究
- 注意事项
- 个人总结
- 分批上样时可以上一个混样,做一个校正,降低分批上样的误差
- iTRAQ与TMT在公司内上标价格相同
- 样品较多时推荐label-free
- 只有PRM可以绝对定量


















评论

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言
查看更多评论
添加红包










