










- 摘要
- 之前写过一篇文章,对clusterProfiler常用注释包进行简单的整理分类2021.05.17【R语言】丨clusterProfiler注释表——KEGG/GO enrich富集图专用_穆易青的博客-CSDN博客。然而在遇到一个原核转录组项目想使用大肠杆菌注释包的时候却遇到了报错,经过一番查阅和测试。终于能够注释大肠杆菌,得到GO/KEGG富集图,在这里将整个过程做个梳理。
- 环境与方法
- R version 3.6.1
- Bioconductor version 3.10
- package:
- clusterProfiler
- org.EcK12.eg.db(E coli strain K12,官方默认包)
- org.EcSakai.eg.db
- 使用问题
- org.EcK12.eg.db与常用的org.Hs.eg.db以及org.Mm.eg.db不同。这个包不能通过require("clusterProfiler")直接使用library(org.Hs.eg.db),需要单独安装。因此我们需要先安装注释包,再进行注释。
- 使用代码
-
require("clusterProfiler") require("topGO") require("Rgraphviz") BiocManager::install("org.EcK12.eg.db") #或者BiocManager::install("ecoli2.db") library(org.EcK12.eg.db) #读取注释包 - GOenrich以BP为例,CC,MF下同
-
ego <- enrichGO( gene = data$V1, OrgDb = org.EcK12.eg.db, keyType = 'SYMBOL', ont = "BP", #CC、MF类型在这里进行设置 pAdjustMethod = "BH", pvalueCutoff = 0.05, #如果图像空白或者富集点较少可以适当调整P值和Q值范围 qvalueCutoff = 0.05 ) write.csv(ego,file = "BP_results.csv") pdf(file="BP_dotplot.pdf") dotplot(ego,font.size=12) #可以补充width和height参数设置长宽,建议MF类型可以适当增加宽度。 dev.off() pdf(file="BP_topGO.pdf") plotGOgraph(ego) dev.off() - KEGGenrich
-
library(DOSE) data=read.table("../diffid.list",header = F) gene<-as.vector(data$V1) gene_list <- mapIds(org.EcK12.eg.db, keys = gene, column = "ENTREZID", keytype = "SYMBOL" ) kk <- enrichKEGG(gene_list, organism="eco", #重点!,这里需要匹配物种简称,查询链接KEGG Organisms: Complete Genomes keyType = "ncbi-geneid", #pvalueCutoff=0.05, #P值和下面的Q值都可以调整,同GOenrich pAdjustMethod="BH", qvalueCutoff=0.05 ) head(summary(kk)) pdf(file="kegg.enrich.pdf") dotplot(kk) dev.off() - 需要注意的是,在我使用的几个注释包里,除了org.Hs.eg.db是要求DE_gene全部大写外,org.Mm.eg.db是要求首字母大写,而org.EcK12.eg.db则是末尾字母大写。当然,如果你的注释文件是官方的,那么格式应该是能够识别的。如果是自己生成的gff文件,那么生成的gene_name最好处理一下。如果格式有问题,程序会反馈提醒
- 结果展示
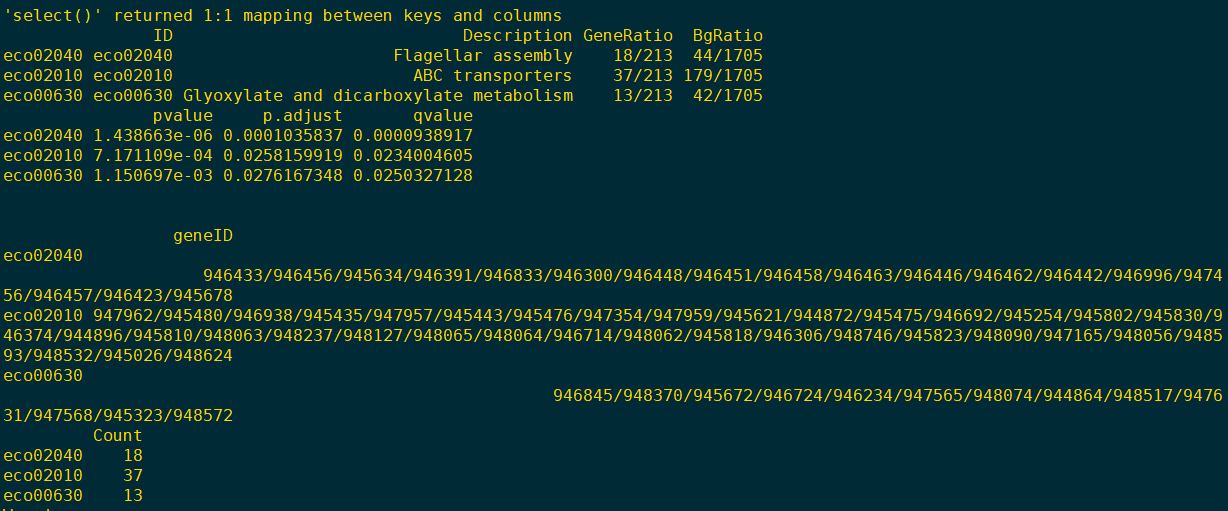
- 使用head(summary(kk)) 命令,如果能够看到富集的ID,并且前缀是eco,那么说明大肠杆菌已经注释成功了。我们再来看看富集图片
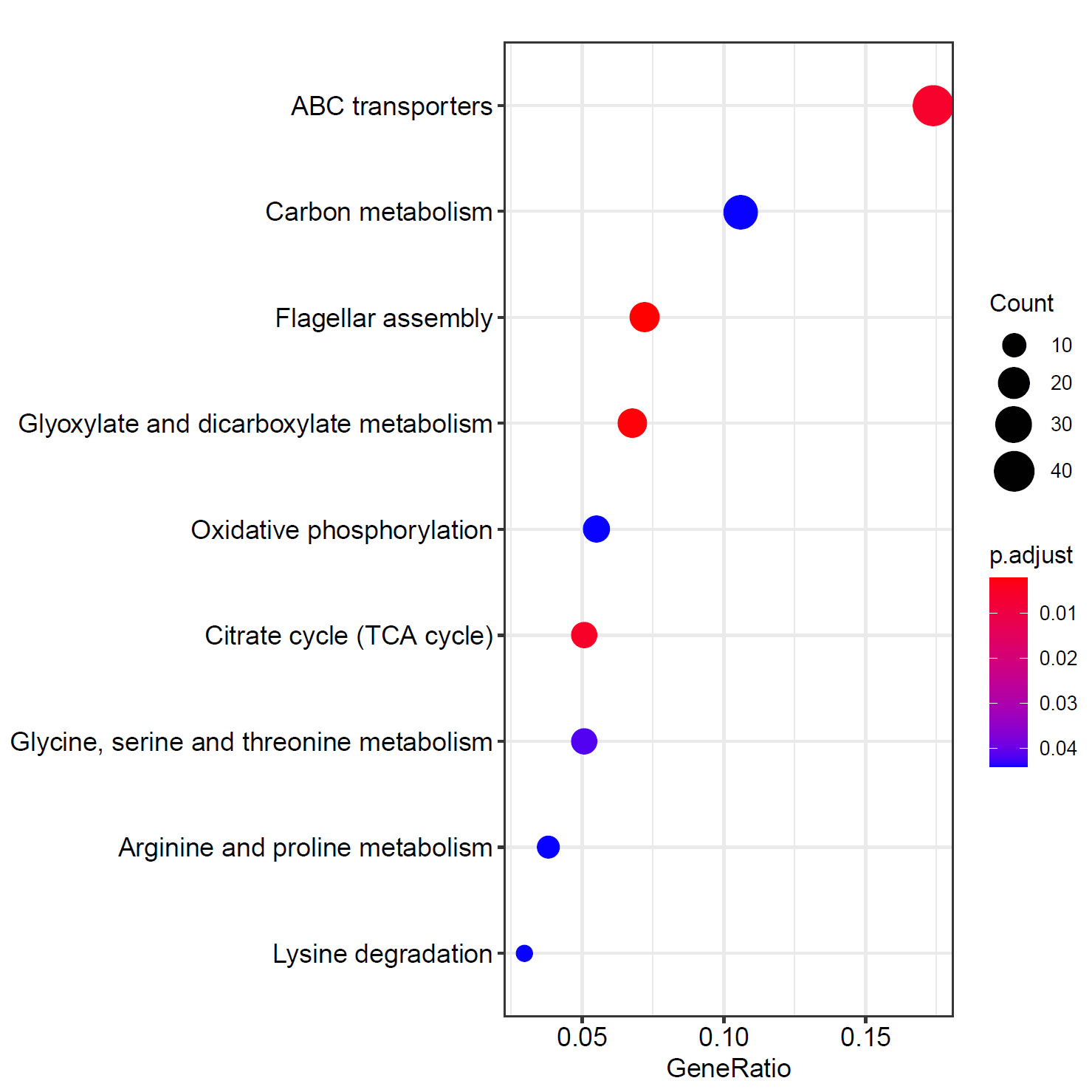
- 这张图我把阈值设置得比较高,所以富集出来得Q值可能超过了0.05
- 总结
- 在梳理RNA-seq的过程中,自己也在不断地了解一些细节。不加思考一味重复,很容易会得到错误的结果.


















 894
894











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











