







零.导读
近几年,蛋白质结构预测领域连续取得重大突破。首先是【AlphaFold】,在可以充分利用共进化信息结合深度神经网络生成空间约束条件并降低相空间的搜索,极大地帮助了蛋白质的结构建模,颠覆了往年需要结合复杂结构采样的算法,现在直接使用能量最小化即可得到预测的结构。随后,在2019年底,David Baker团队发表了【trRosetta】,其集合深度学习的诸多进展,并与Rosetta建模软件结合,使得预测蛋白结构的门槛大大降低(在笔记本折叠蛋白) 。在【trRosetta】的文章中, 作者还发现了一个有趣的现象,对于很多之前设计的de novo design 的人工蛋白,在没有同源序列(MSA)的情况下,只凭单序列输入就可以预测到比较可靠的结构。
这个结果似乎暗示,trRosetta模型不但学到了用共进化信息来推断空间约束,也学会了某些序列和结构之间的本质关联。于是作者提出了两个问题,
1. 这些信息能否用来生成与训练集序列不相关的新蛋白?
2. 对于给定的空间约束(结构),模型能否通过反向传播优化序列,也就是实现“design”的操作?
一、引理
蛋白质的结构和序列之间的关系,可以用条件概率和贝叶斯公式表示
P(seq|struct) = P(seq,struct)/P(struct) = P(struct|seq)*P(seq)/P(struct)
其中P(struct|seq),给定序列求结构,是trRosetta解决的结构预测问题。P(seq)是序列与结构无关的概率,也就是天然蛋白中氨基酸的频率。P(struct)是与蛋白序列无关的结构信息,即背景。在文章中,作者对背景噪音单独训练了一个神经网络,神经网络的结构和trRosetta相似,但输入的MSA为只与蛋白长度相关的随机噪音。
二、不给结构随便幻想
有了这个简单的概率模型,作者的第一个问题就是如何让神经网络去幻想(hallucination)新的蛋白结构,我们能否随机在P(seq,struct)中找一个位置然后找到它附近的极值?这就要说到深度学习中的一个有趣的问题DeepDream(深梦)。
DeepDream
这个方法反映的是一个神经网络是怎么“认识”世界的,当你训练好一个图像分类器后,输入一张图片,deepdream就在图中拼命寻找符合它所认识物体的pattern并加以放大,最后得到一张非常魔幻的照片。

这也正是幻想蛋白希望得到的效果,如果给定一个条序列(比如一条随机序列)时, trRosetta预测出来的空间约束往往是缺乏特征的。如果能够像deepdream一样在里面寻找像是理想蛋白的特征并加以强化,最后我们就能得到一个具有良好蛋白性质的空间约束及其序列。
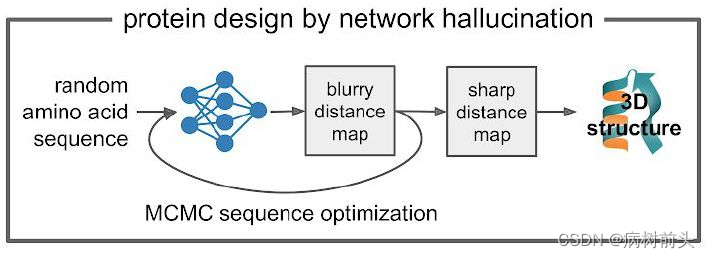
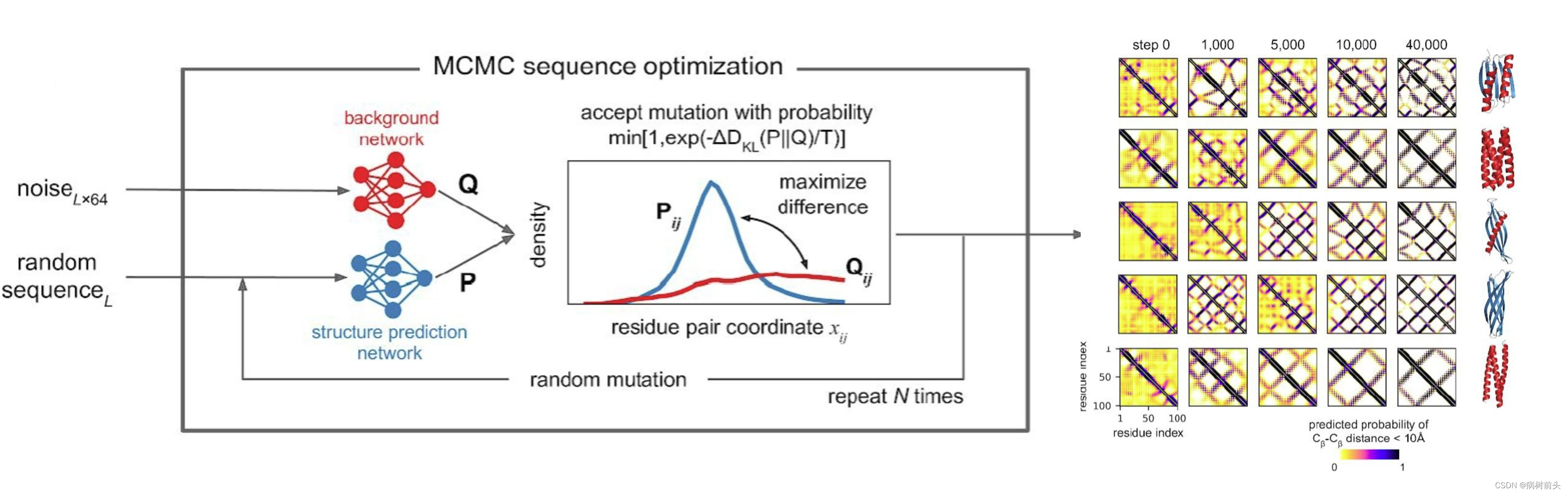
MCMC序列优化
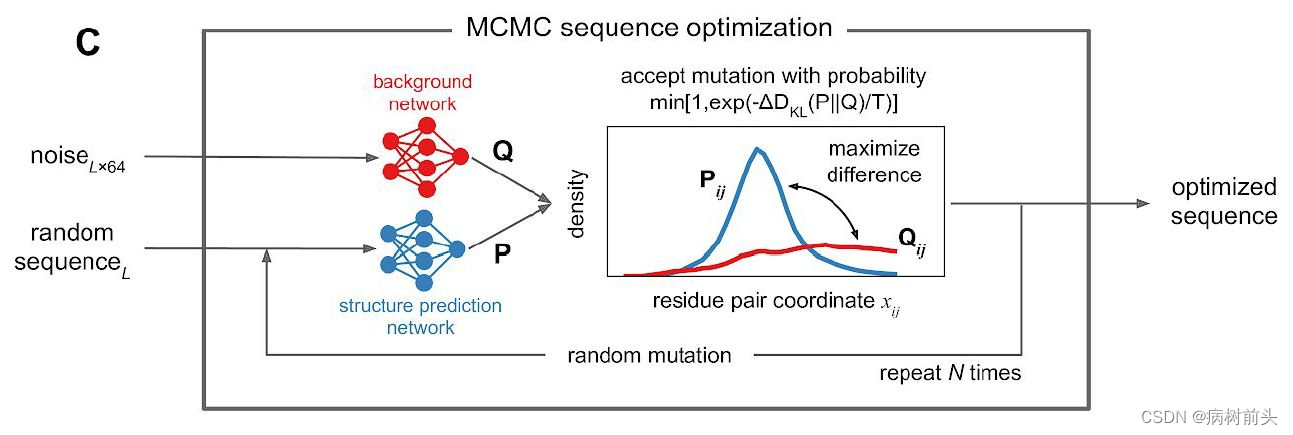
具体的做法也相当简单,首先给定一条初始序列(可以完全随机也可以是有意义的序列),接着将一个大小为Lx64的随机噪音输入背景网络,得到背景的空间约束。然后从初始序列出发,通过trRosetta网络预测其空间约束,最初的约束可能分布相当弥散(因为序列不具有明显结构特征),计算这个分布与背景分布的差异,如果两者的KL散度越大,则说明得到的空间约束越像一个蛋白。作者在序列中随机引入点突变,用Metroplis判据来不断优化(增大)KL散度。
通过一个模拟退火的过程,随着温度不断降低,作者得到了与噪音相比,KL散度非常大的序列。从D图中可以看到,天然蛋白,从头设计的蛋白,以及模拟退火后的幻象蛋白序列与背景空间约束分布的散度依次提高。40000步后幻想序列具有非常高的KL散度。
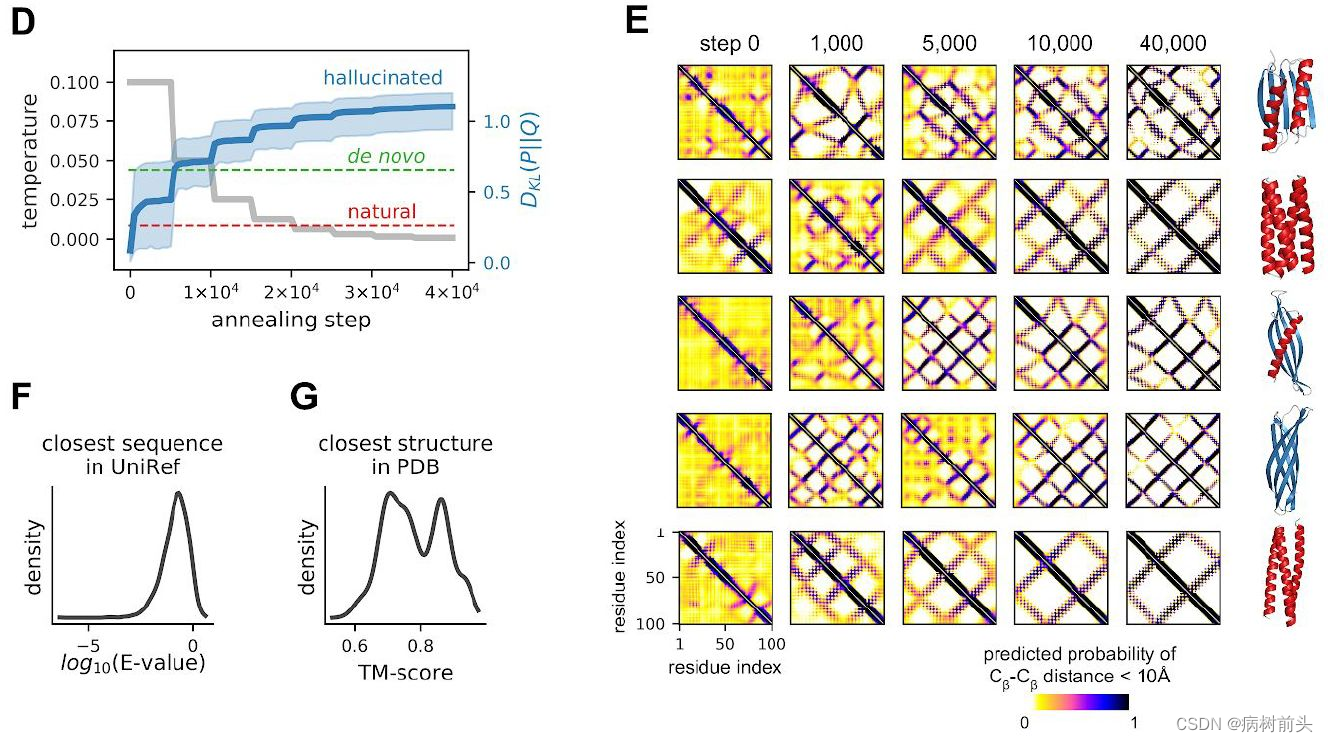
再比较一下序列,会发现幻想出来的序列距离天然蛋白非常远,但结构上却在PDB中具有很好的匹配程度。也就是说,幻想出来的蛋白是序列独特但结构老套的蛋白。当然这并不奇怪,PDB库早已经被报道覆盖了蛋白质大部分可能折叠的空间,更何况trRosetta就是基于PDB结构进行的训练。最终作者展示了多种全新幻想出来的序列,形成覆盖全α全β或两者混合的各类拓扑结构。
三、给定结构幻想序列
蛋白质设计的目的则在于优化P(seq|struct)。而在第二章节的MCMC采样过程中,每次随机突变一个氨基酸的方法效率较低,并没有用到深度学习的关键技术“反向传播”。这个方法可以使我们根据目标分布与预测分布的差异,有目的地批量更新氨基酸,为了用连续变量编码氨基酸,文中采用PSSM来描述一条序列。这套方法被称为trDesign。
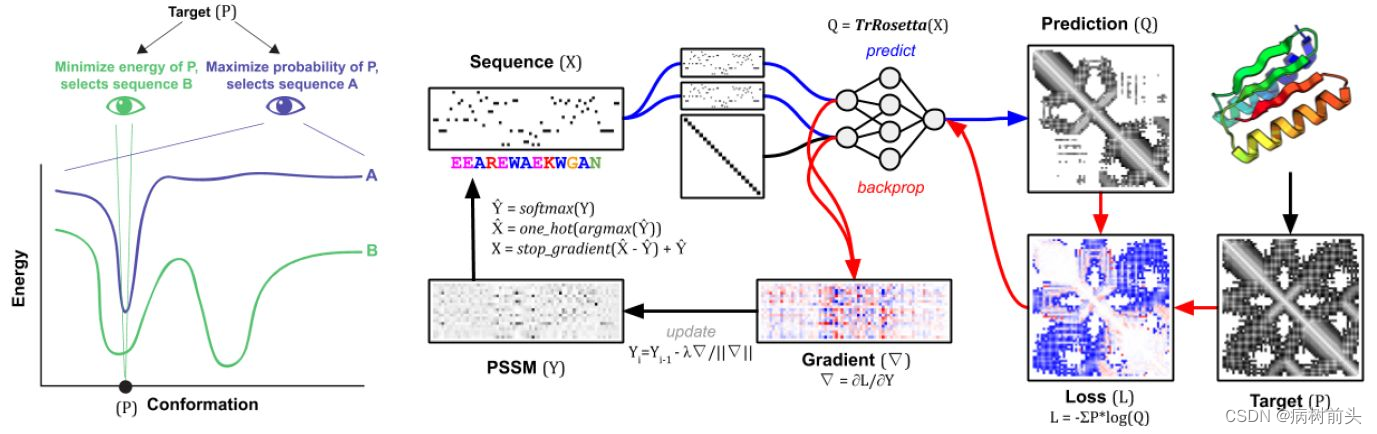 当我们有了一个目标结构的时候,就可以计算目标的空间约束(Target-P),用一条随机序列通过trRosetta可以得到预测空间约束Q,P和Q的散度,就是我们希望最小化(使得Q接近P)的损失函数(注意在幻想时是希望最大化和背景的差异)。
当我们有了一个目标结构的时候,就可以计算目标的空间约束(Target-P),用一条随机序列通过trRosetta可以得到预测空间约束Q,P和Q的散度,就是我们希望最小化(使得Q接近P)的损失函数(注意在幻想时是希望最大化和背景的差异)。
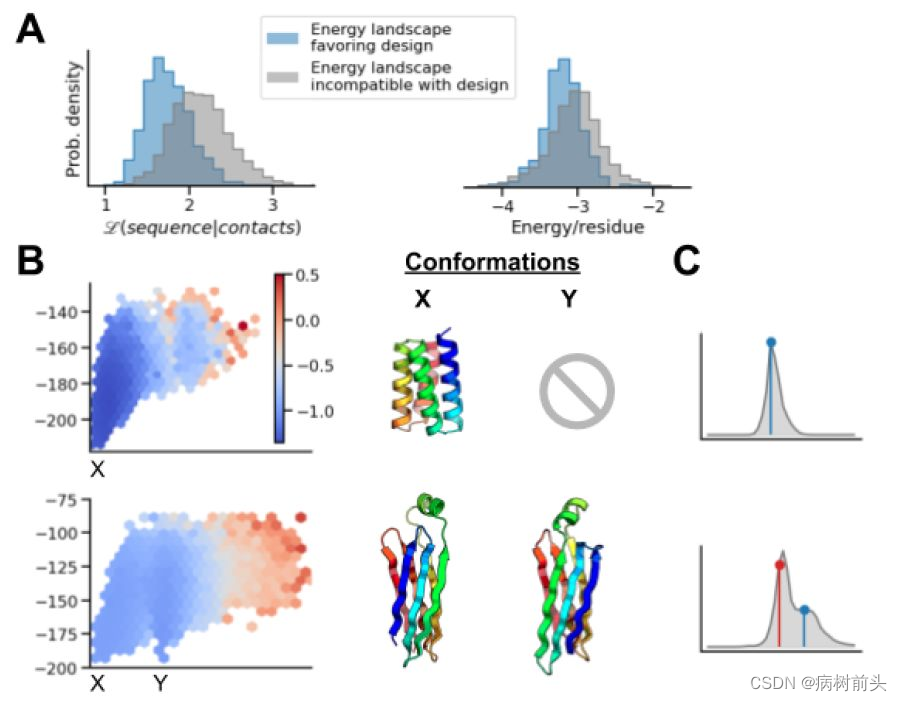
这个流程可以快速的对输入序列进行优化,只需几十步的迭代就可以得到收敛的结果。通过对Foldit玩家设计的几千个蛋白进行分析,传统的势能面打分Pnear可以得到与实验较好的关联性,但开销十分巨大。而trDesign的损失函数与Pnear有很好的关联性,且对实验验证成功的例子有更好的区分度。说明trDesign所优化的是整个能量面,即降低目标构象的能量同时提高其它构象的能量。但缺点在于对native结构的优化不如Rosetta深入,这主要是受限于模型的精度。
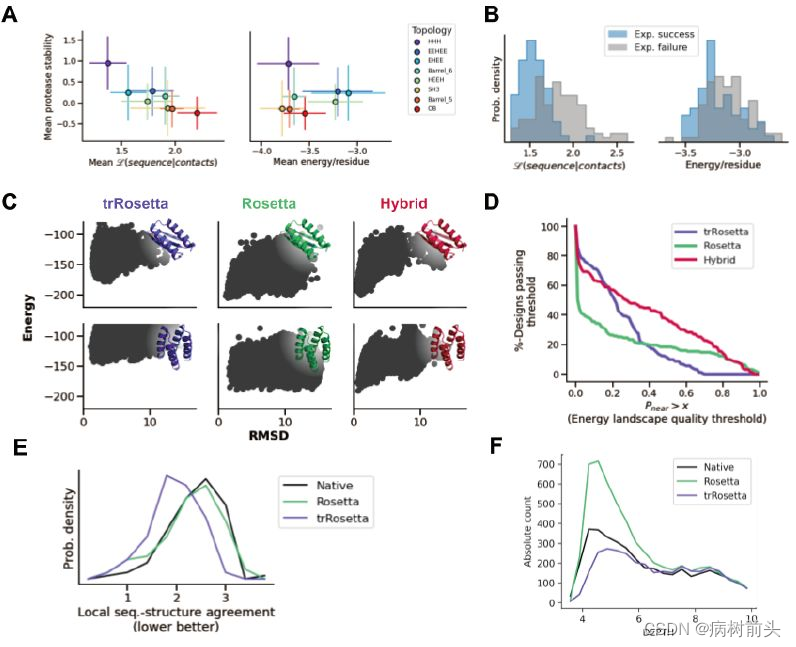
那么究竟trRosetta学到了什么呢?作者列出了三点:
1. 一些距离的双峰分布说明模型学到了全局或二级结构的不同堆叠状态;\
2. 相对天然蛋白或De novo设计的蛋白而言,trRosetta设计的蛋白具有更理想的局域序列-结构关系(图E);
3. trDesign设计的蛋白具有更少的表面输水侧链(图F),尽管它们对全局最小影响可能不大,但如果表面过多的疏水侧链,这可能使得蛋白质会偏向折叠成使这些氨基酸包埋的亚稳态结构。
总结: 作者开发的这套方法不但可以进行快速的蛋白质设计,而且其效果是可以优化整个能量面的形状,如果与适合在局部深挖的Rosetta FastDesign相结合,可以达到远超原来蛋白设计流程的效果。
刘源:有人可能会说,你能想象出新蛋白又有什么用?能反向传播又如何,不就比design快点么?于是重点来了,由于这两个方法都是基于trRosetta模型来的,所以如果合理的设计损失函数,人们就可以做到固定一部分想要的结构,然后幻想生成剩下的部分!这也就是深度学习里常见的inpaint问题,挡住一部分图片,自动补全新图。而且最终得到的设计是非常接近ideal的稳定蛋白,具有很多好的性质,从这个角度trDesign可以看成是一种滤镜,给输入的蛋白“磨皮”,让它更加完美(稳定易折叠,优化能量面)。最近的一个例子是Baker组设计的一个IL-2的mimic从头设计蛋白【neo-2】,在文章中作者使用了大量复杂的算法生成主链构象再进行设计,而理论上这个操作可以在新的框架中一步到位。相信这套方案在成熟之后会在设计抗体、疫苗等重大问题上带来突破性进展。
皮卡车:这两篇文章的idea是如何出来的,和Sergey的一些经历和想法分不开。Sergey 主要做的是共进化相关的工作,GREMLIN为主,也可以叫markov random field, potts model,self-supervised learning等。通过对MSA的分析来得到蛋白质的接触图谱。然后遇到的一个问题是,目前的结果是通过分析单层神经网络的参数获得的。如果层数增加,物理意义不明晰,就在模型中丢失了接触图谱的解释性,于是他搞了一套基于梯度的分析方法,Seqsal,把输入当变量,就可以从多层神经网络中,得到蛋白质的接触图谱,于是各种模型,autoencoder,VAE等都可以通过这个方法来重新解析。在后来,trRosetta有了,是一种从序列到结构的分析方法。那么倒过来把序列当变量,通过调整序列来降低模型的损失函数,同时又把序列推离序列噪音,deep network hallucination就出来了。在损失函数中增加一项给定的结构约束,trDesign就出来了。在有了这些想法之后,能够在几个月时间内快速推进算法和实验。除了baker实验室,其他地方也难找了。
1. 前言:
随着alphafold2突破性预测蛋白结构的成功,学术界也开始尝试探索如何使用它进行高精度的蛋白序列设计。
2. 计算方法:
计算方法:
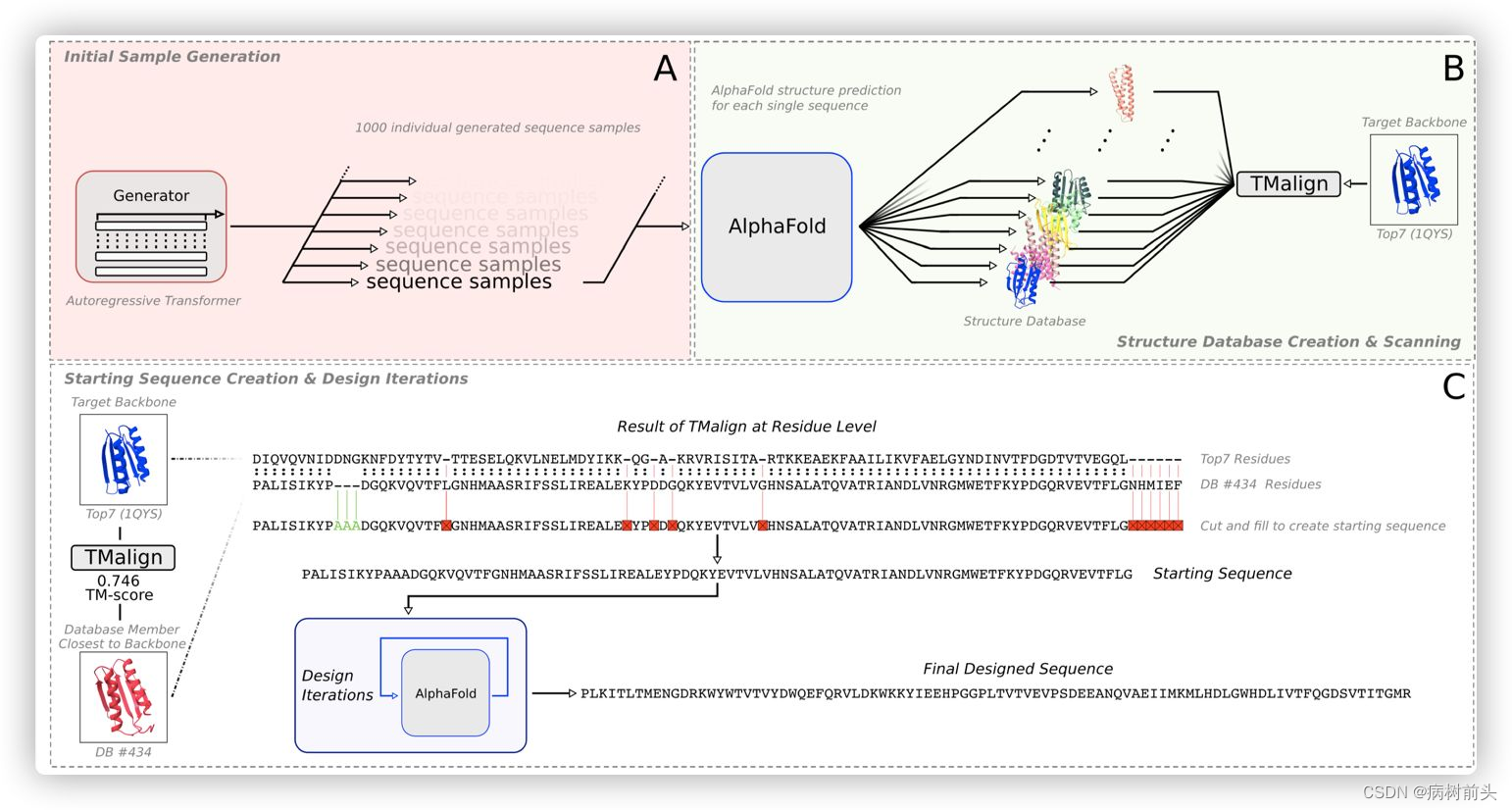
序列开始并非使用随机模型, 而是使用了一个自回归的transformer来生成初始的序列。大概1000条denovo的序列(图A)。
将1000条序列喂给AlphaFold预测所有的结构(relaxed,最高pLDDT的模型)进行保留。随后使用TM-align将target的backbone和de novo设计的序列结构进行对比(图B)
将最高的Tm-score的结构的序列作为初始父代序列,并且只保留aligned结构motif部分的序列,没比对上的用丙氨酸进行替代(图C)。
经过这样处理之后,将预测对的residue fragment提取了出来,比随机生成更有利于序列空间的搜索。
迭代end-2-end设计
设计方法的核心是通过贪心半随机的算法对序列空间进行采样,接着使用AlphaFold预测结构,直到生成与目标结构的backbone尽可能地相似。首先同样采用了distance map loss的计算方法,来比较设计的结构与真实结构之间差异:
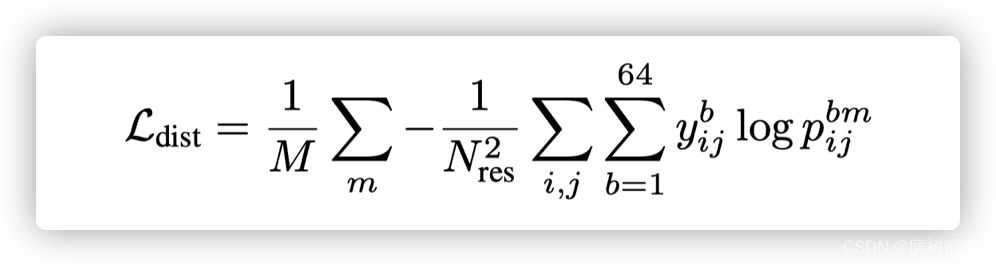
ij是每个氨基酸对, y是真实的距离分布特征,p是预测的距离分布特征。在推断过程中还计算每个残基的pLDDT,然后在5个参数集上取平均值,但不在序列长度上取平均值。
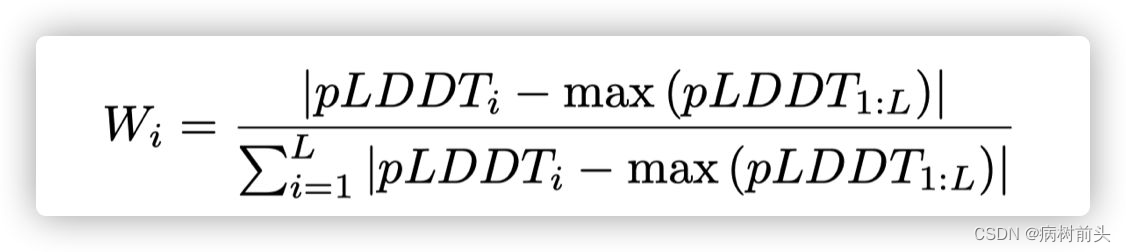
这个weight被用于设定为序列采样的概率。假设pLDDT高的区域,氨基酸是稳定的,突变时将选择概率低的区域进行突变。
一旦决定了哪个区域的氨基酸应该被采样后,这个位点将随机等概率突变成另外一些氨基酸的类型(除了cys)。并且当这个突变让distance map loss降低时(改善预测结构吻合度时),保留此突变。最后通过如此迭代了20000轮突变,distogram score收敛。
Fast AlphaFold inference


3. 设计结果
作者使用了人工设计的Top7作为测试集
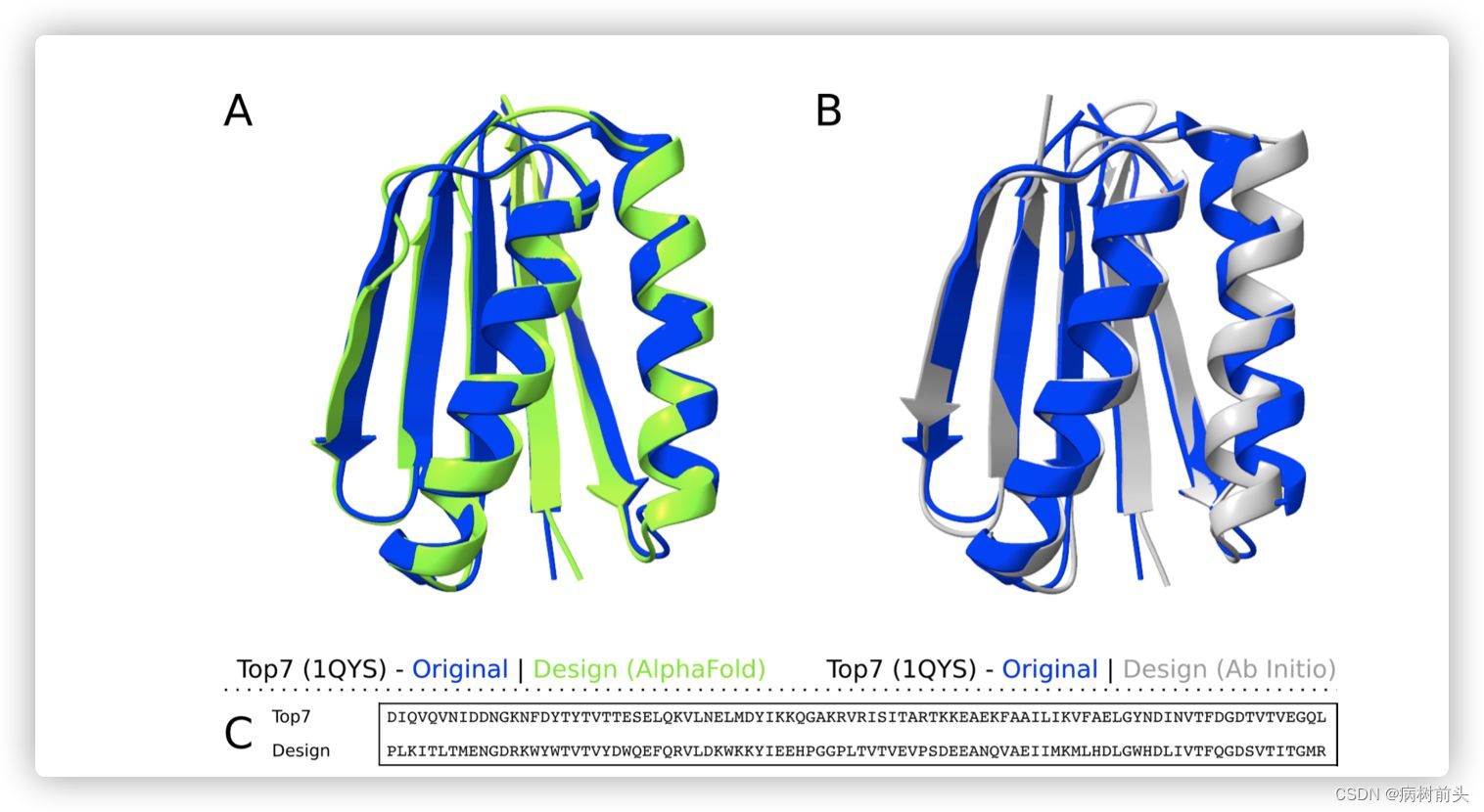
在第一阶段进行序列设计时,af2预测的TM-score仅有0.746,经过上述的方法进行迭代设计之后,新设计的序列与Top7的相似性仅为27%。将此序列使用af2验证时,全局的RMSD仅为0.736 Å,pLDDT score为91。而使用trRosetta进行预测时,Cα-RMSD为2.637 Å,TM-score为0.679。第三种检查方法为ab initio fragment-based的方法进行预测,经过15000个采样后,最好的结构Cα-RMSD 为1.279 Å。均证明,设计的序列与目标结构可能是同一种Fold。

Top7设计成功后,作者进一步尝试设计未在训练集中的数据Peak6 (PDB ID 6MRS)、Foldit(PDB ID 6MRR)、Ferredog-Diesel (PDB ID 6NUK)。初始序列对应匹配TM-score为0.596-0.7之间,经过设计后,af2预测结构的Cα-RMSD降低至1Å以内,pLDDT score > 85。使用ab initio fragment-based的方法进行预测Cα-RMSD均小于3Å。设计的序列与目标模板序列相似性均低于30%。在多种结构预测工具中,trRosetta预测的结构Cα-RMSD较大,这可能与输入MSA质量较差有关。
4. 讨论
作者通过使用缩水版的alphafold2进行fix-backbone设计,本质上即使用基于pLDDTscore版本的mcmc序列采样,最后通过结构验证所设计的序列可靠性。此设计方法中没有使用到能量函数的概念,因此推测AlphaFold已经学会了一些与能量功能相关的蛋白质结构的泛化。
最近AlphaFold2的消息刷爆全网,以超高的预测精度吸引了无数结构相关从业人员的眼球。里程碑式的事件表明Deep learning是protein design未来必须走的一条路,并且谷歌已经在走了。在Protein design领域,David baker(RosettaCommon)一直走到在世界的最前沿。在今年年初,笔者我发过一篇基于人类想象构建蛋白质拓扑结构的设计方法(Topobuilder)。对,没错。在新的trDesign Hallucination的思路下,已经过时了。不禁感叹,技术变化迭代之快。
1. Why Hallucination
Hallucinating在今年的8月份闪亮登场,其收到了DeepDream的启发,将反向传播/MCMC的方法应用在Protein Design/Generation中。(*https://zhuanlan.zhihu.com/p/185073475)
Hallucination其基本原理是通过MCMC/Gradient descent对trRosetta的input的几率序列矩阵进行采样,不断优化(增大)与背景噪音分布的差异,得到有显著蛋白特征的Contacts Map,再基于这些一系列的6D Geometric Feature使用trRosetta生成目标蛋白骨架结构。
近日,这个框架有了新的特性(也是兑现了第一篇文章预印时立下的flag),那就是在Hallucination的过程中引入不连续的功能性的Motif constraint,换言之,你只需要输入一段有功能性的蛋白质结构,剩下的就交给trRosetta自行生成。
做denovo protein design的同道们可能都知道,在年初Topobuilder(https://zhuanlan.zhihu.com/p/113435234) 巧妙地结合人类直觉,设计出了RSV亚单位表位的鸡尾酒疫苗,吸引了许多免疫学家的关注。其最大的亮点在于可以将不规则的结构表位引入到新的denovo scaffold中,并成功引起了动物体内的免疫反应。
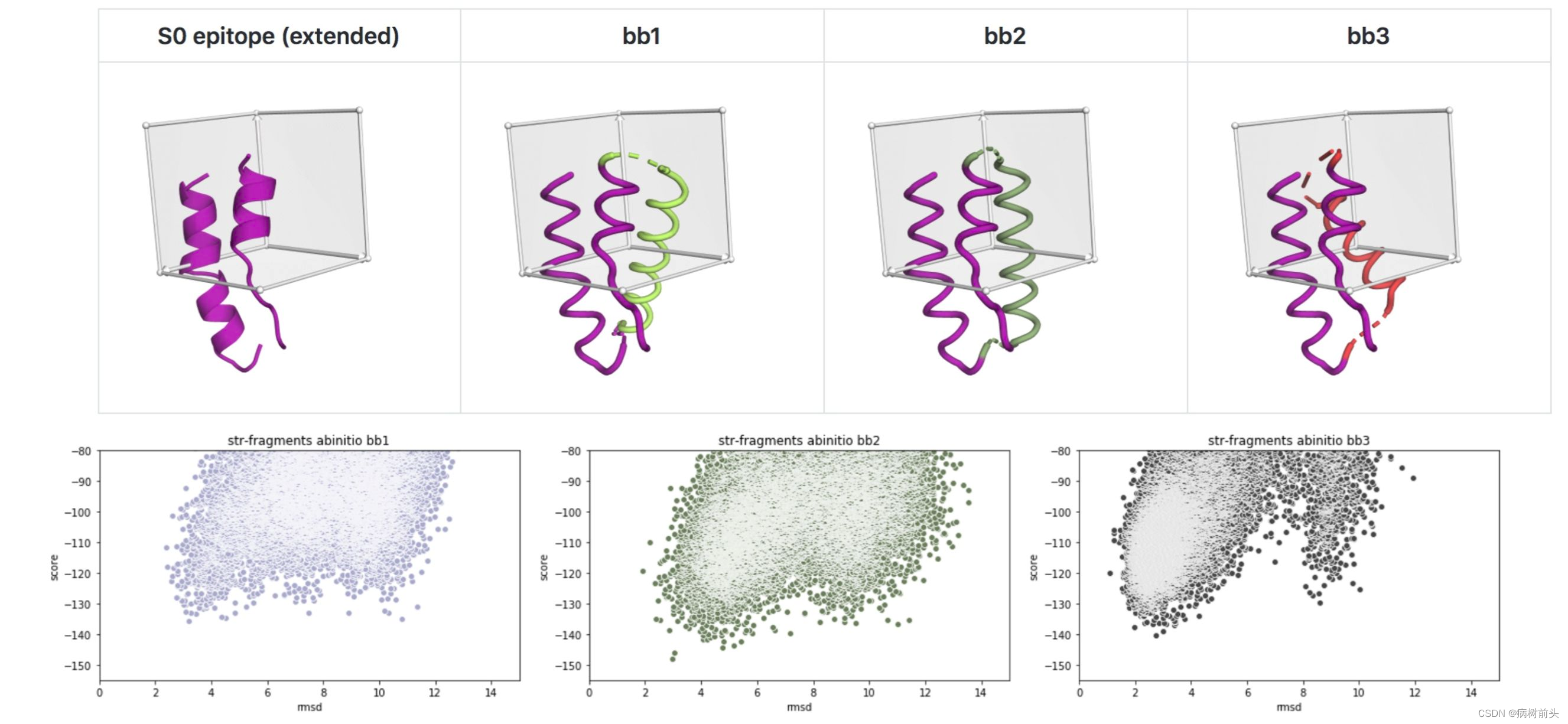
Topobuilder的优势在于人类直觉,但缺点也在人类直觉上。当我们遇到的表位比较复杂时,人的大脑就无法很好地想象出一个结构来适配它。但这在这类问题,Hallucination框架下,可以被轻而易举地解决。
2. How to Hallucination
Tischer等在原先的Hallucination思想上,增加了引入Motif表位的概念。在新的论文中,主要的亮点在于解决如何在Hallucination中将Motif放置在蛋白质序列空间中的问题,以及如何控制trRosetta幻想出特定motif的结构特征。
1. 先来说说如何控制trRosetta幻想出Motif结构特征。
关键的点在于如何定义反向传播时的Loss function,作者通过将Motif和scaffold部分的Loss进行分开定义为motif satisfaction (MS) loss 和 a free hallucination (FH) loss。MS负责控制Motif区间的序列概率梯度方向,使得在每次迭代尽可能使这个区间的结构特征向我们需要的Motif方向发展。而其他起到支撑作用的序列尽可能和背景分布不同。通过这混合的Loss function,即可定向优化生成蛋白质的结构和序列。
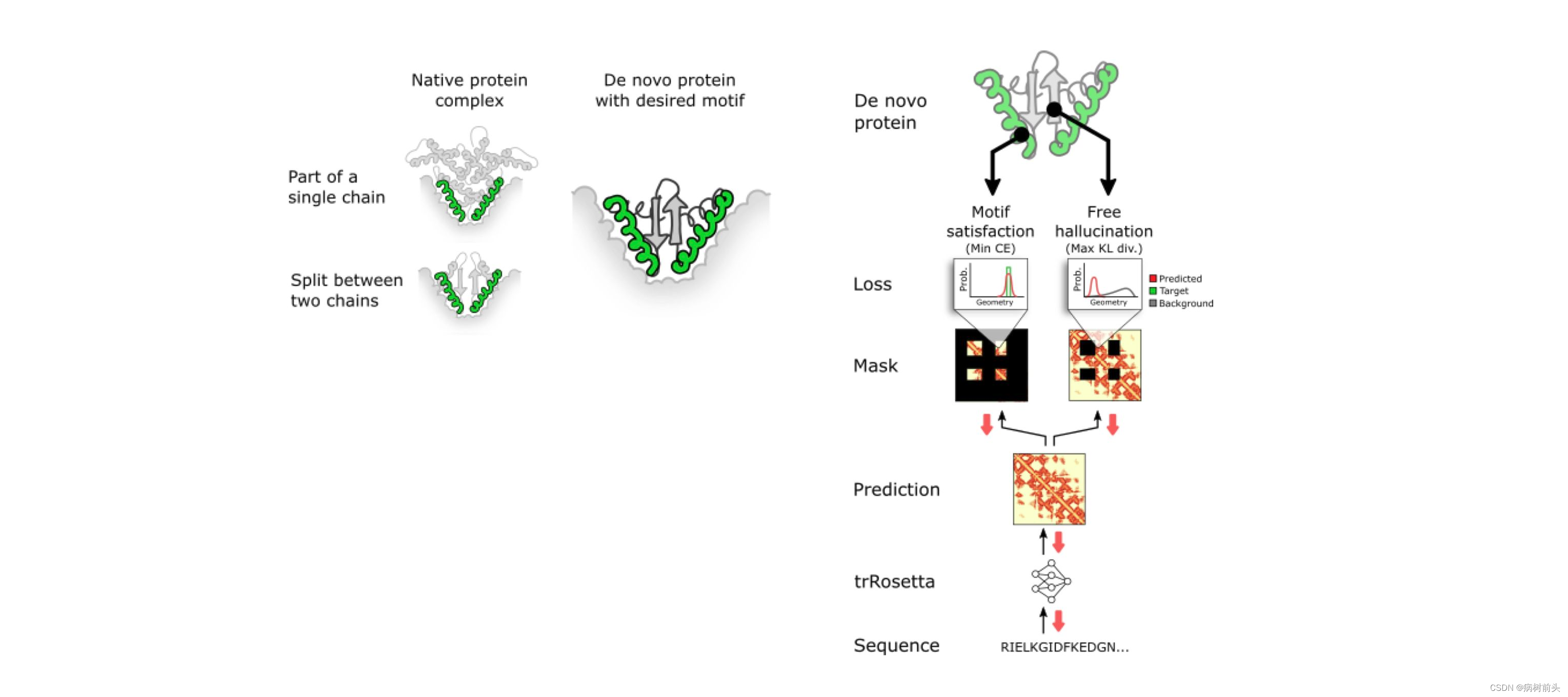 2. 再来说说如何控制Motif的插入位置。
2. 再来说说如何控制Motif的插入位置。
作者主要使用了两种方式:
通过人定义的方法进行Hallucination
通过卷积交叉熵和贪心算法自适应确定Motif的位置
第一种方式简单粗暴
就是人为定义Motif/Regions的区间,生成许多不同Regions的长度序列组合,每个组合进行30000步的MCMC,模拟了大量的Hallucination轨迹,这种做法。很Rosetta。虽然思路简单,但是效果好!对于简单的不连续Motif,这种搜索方式可以更好地对Loop区间长度进行采样。
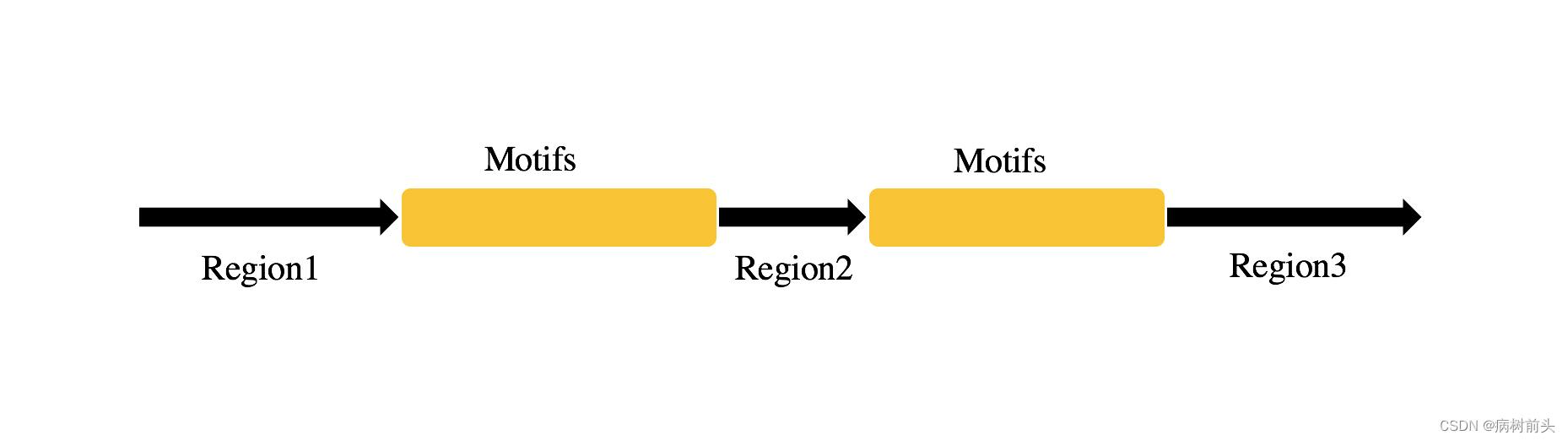
第二种方法就有点"卷"了
与第一种方法不同,我们搜索的序列的长度是固定的。对于复杂的motif(如混合α/β-loop的Motif),通过人的想象就很难定义Motif区间的位置,大量的试错效率十分地低,因此作者使用了一种更加聪明的方式来Place这些Motifs。
算法的原理是在trRosetta的6D Geometric的Feature Maps上进行遍历的卷积,求得交叉熵Feature Map(为什么是算交叉熵,因为trRosetta预测出来的结果是一个概率分布),最后使用贪心算法来确定,我们需要在哪个区间范围内进行MS/FM的loss计算。本质上就是在找trRosetta预测出来的结构特征中,哪个区域和我们的target最相似,然后我用这部分的Feature去求Loss。
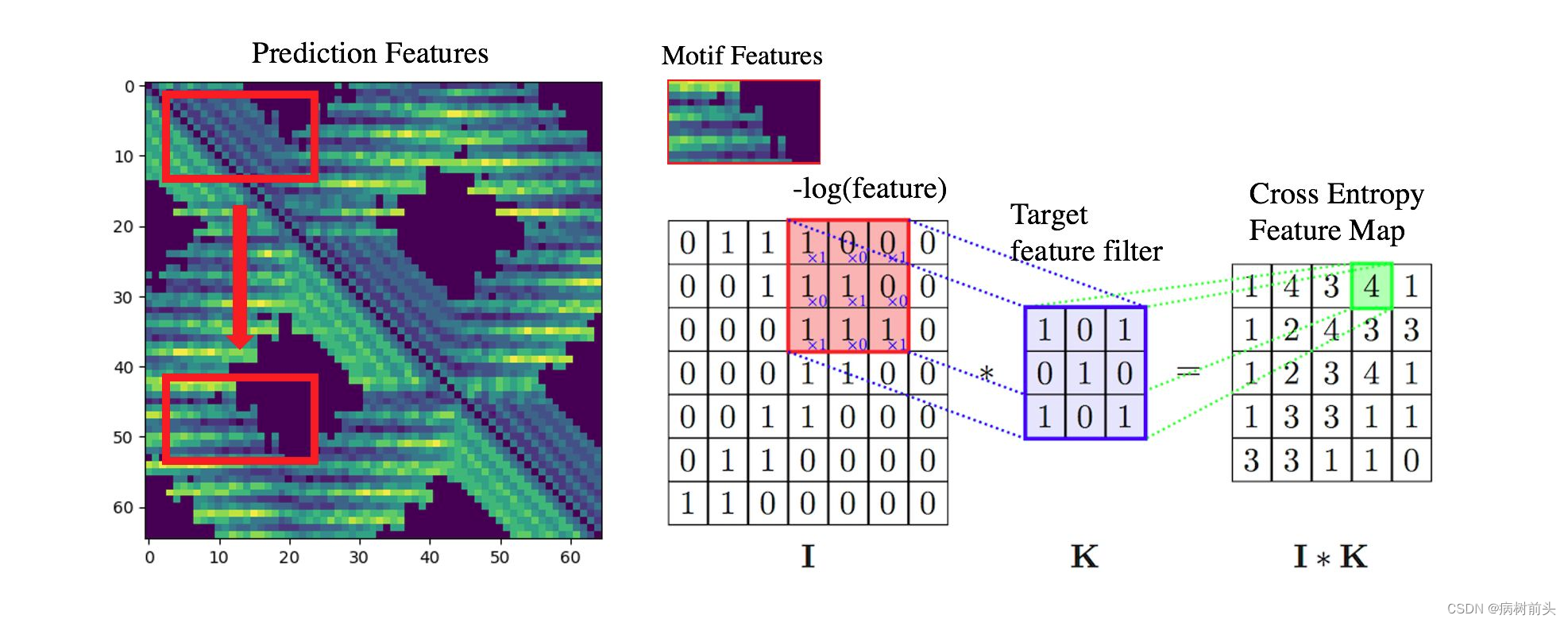 具体的操作步骤:
具体的操作步骤:
在第一步迭代时,随机将Motif的区间进行定义,进行第一轮Hallucination。
在第二次迭代开始时,trRosetta会对上一轮输入的序列进行预测给出Prediction的Feature,再此结果的基础上,进行上述的卷积求得交叉熵Map,使用贪心算法(每次都选交叉熵最小的就是在motif最相似的区间)。然后反向传播求最开始的序列概率矩阵的梯度。并更新输入的序列。
如此往复,在求Loss之前先“卷”一遍。随着迭代次数的增加,被Mask的Motif区间就会慢慢地固定下来在一区间内。
这套算法下来最大的好处就是,机器可以自己确定我应该在哪个地方放置我的motif。
Ps: 当然也可以通过尝试多种序列长度,与第一种方法本质上殊途同归。
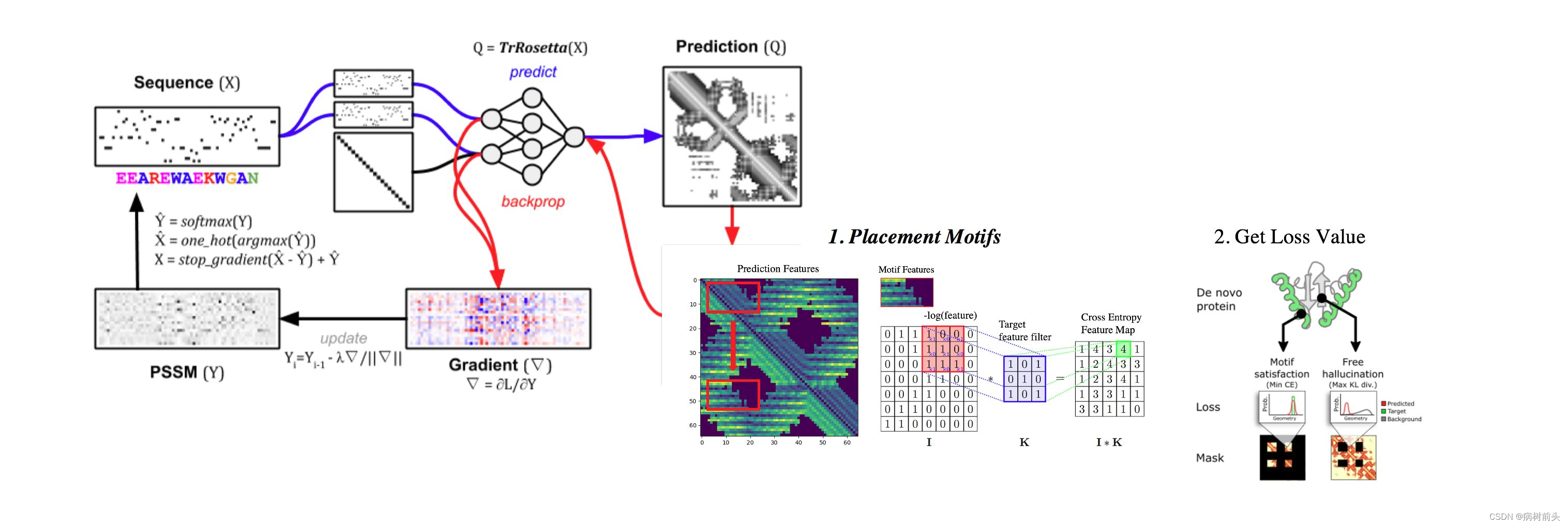
3. Proof of concept
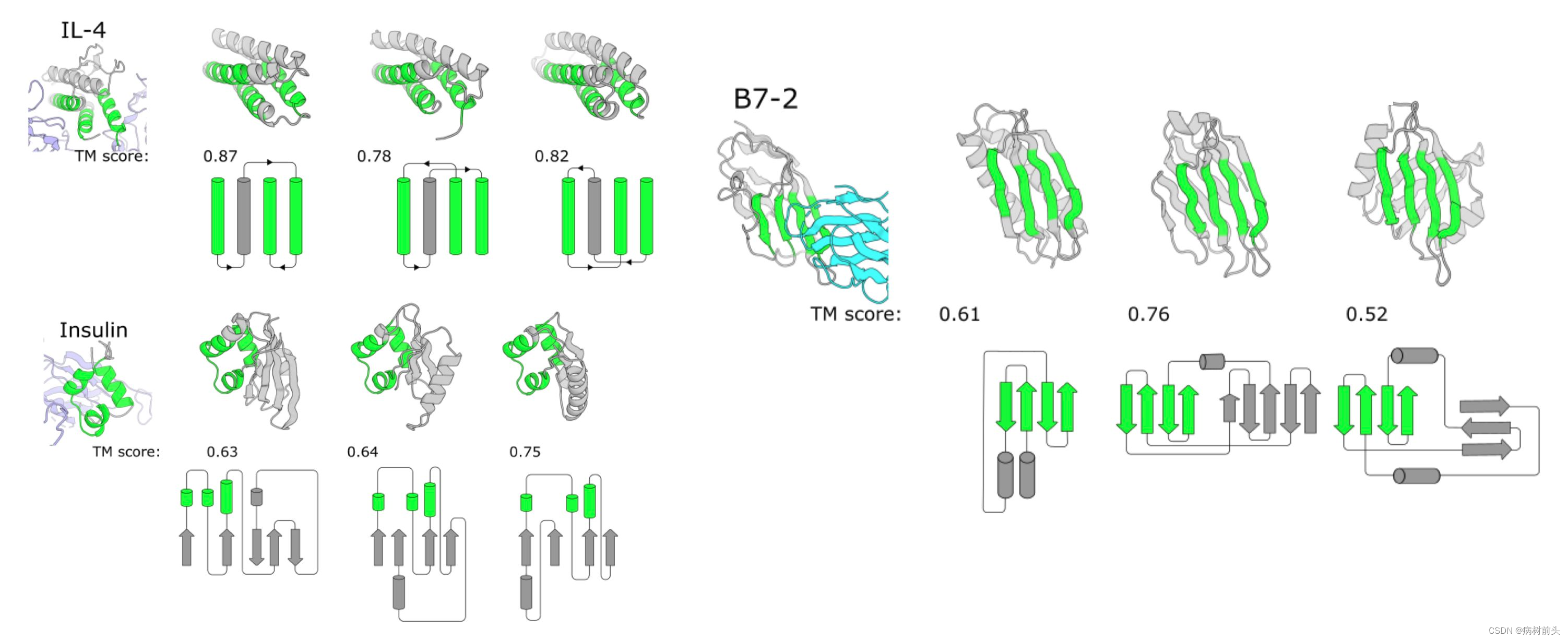
有了新的框架,作者对好几个Motif进行定向Hallucination的测试。作者在多种Motifs类型上做了尝试。。(强大无比)许多的Motifs都能找到与之适配的scaffold骨架。
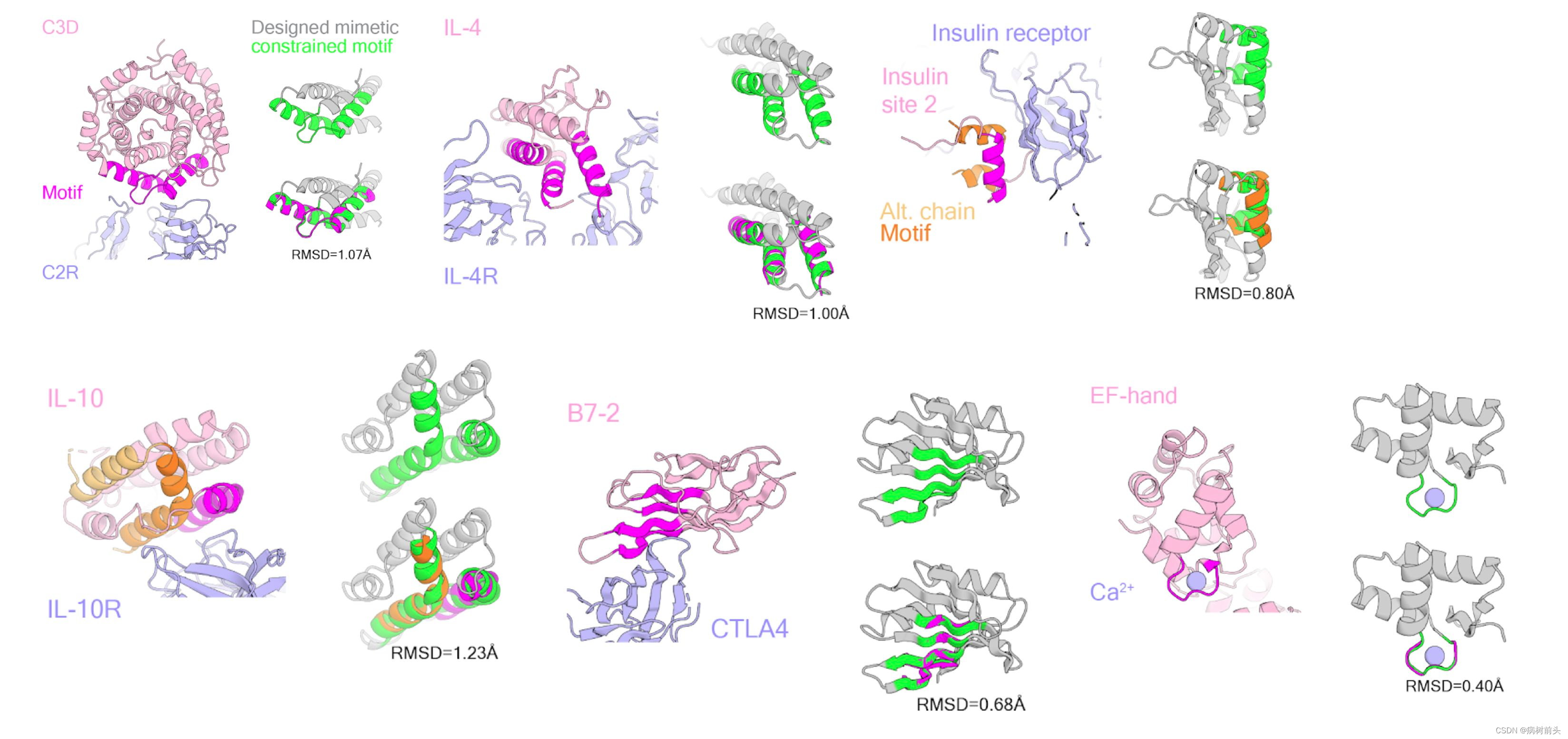 除此以外,多次Hallucination的结果也表明,同一个Motif是可以被各样的Scaffold所适配。(温馨提示:您的GPU机时已不足)
除此以外,多次Hallucination的结果也表明,同一个Motif是可以被各样的Scaffold所适配。(温馨提示:您的GPU机时已不足)
End
毫无疑问,BakerLab将DeepDream的思想很好地实践并融入到了ProteinDesign这个领域并获得成功。这次的进步主要是颠覆了Functional de novo design中模板缺乏的问题。在未来,trRosetta的设计/预测精度再继续提升时,我们就能真正地实现”一键蛋白质设计“了。
Rosetta TopoBuilder: 基于参数的Denovo Scaffold生成器
零、前言
在RosettaCon2018大会上TopoBuilder由Bonneau实验室提出,为了克服非规则Motif结构难以搜索到可移植的Scaffold的问题。TopoBuilder是很好的创建理想化拓扑结构的工具,具有多个维度进行拓扑定义,并且可以通过二级结构之间的距离进行Loop搜索,遍历所有的组合形式,来产生Denovo的Scaffold。该算法已成功应用在鸡尾酒法疫苗设计当中。
一、基本概念
TopoBuilder中有Layer、Config和Motifs三层控制的属性:
- Layer属性
图层概念,将蛋白的以Z轴作为切割平面,将3D结构分为若干层。如下图右侧中,我们通过俯瞰蛋白质的结构,5条β折叠(红色)横着排列,两个α螺旋(蓝色)也是并列排列。从中间切开就形成了2个layer层了。由此可以理解左侧两个子图为每层结构的平视图。
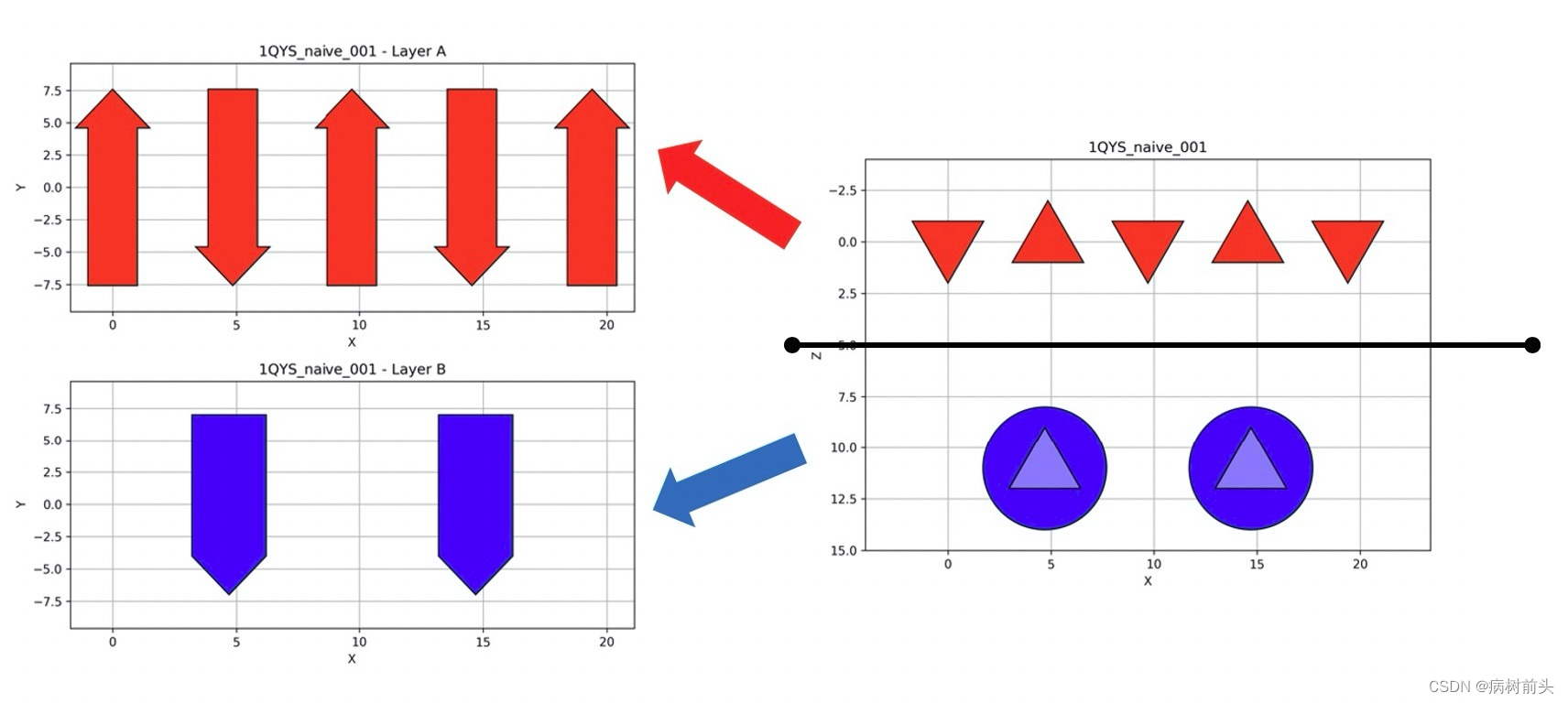
Layer中应该定义好每层二级结构的长度和类型以及自定义调整:
type: 二级结构的类型,H为α螺旋,E为β折叠;
length: 二级结构中包含的氨基酸数量;
==ref: 该字段负责引入Motif的信息; ==
shift_y(z): 自定义某个二级结构在y或z方向上的平移,如对某个螺旋结构进行调整方向为y轴方向+2,那么将会使得该结构平移“突出来”
shift_x: shift_x的操作逻辑略有不同,如果shift作用于该层layer的第一个二级结构时,对整一层进行平移,如果作用在第二个二级结构时,代表该结构从第一个二级结构为原点进行左右平移x个埃的距离。(这个比较坑)
tilt_x(yz): 定义每个二级结构在xyz空间内的倾斜度

使用[ ]字段代表一个layer,{ }字段定义一个二级结构的信息。从上述的例子中定义了两个layer。每个layer中包括2个螺旋 ,每个螺旋的长度为16. 其中第二个layer中的第一个螺旋为我们需要移植的Motif结构,具体的结构由ref字段进行定义。
2. Config属性
定义全局拓扑结构的参数(二级结构之间的距离),如图所示:(通常这些属性是有默认值的)
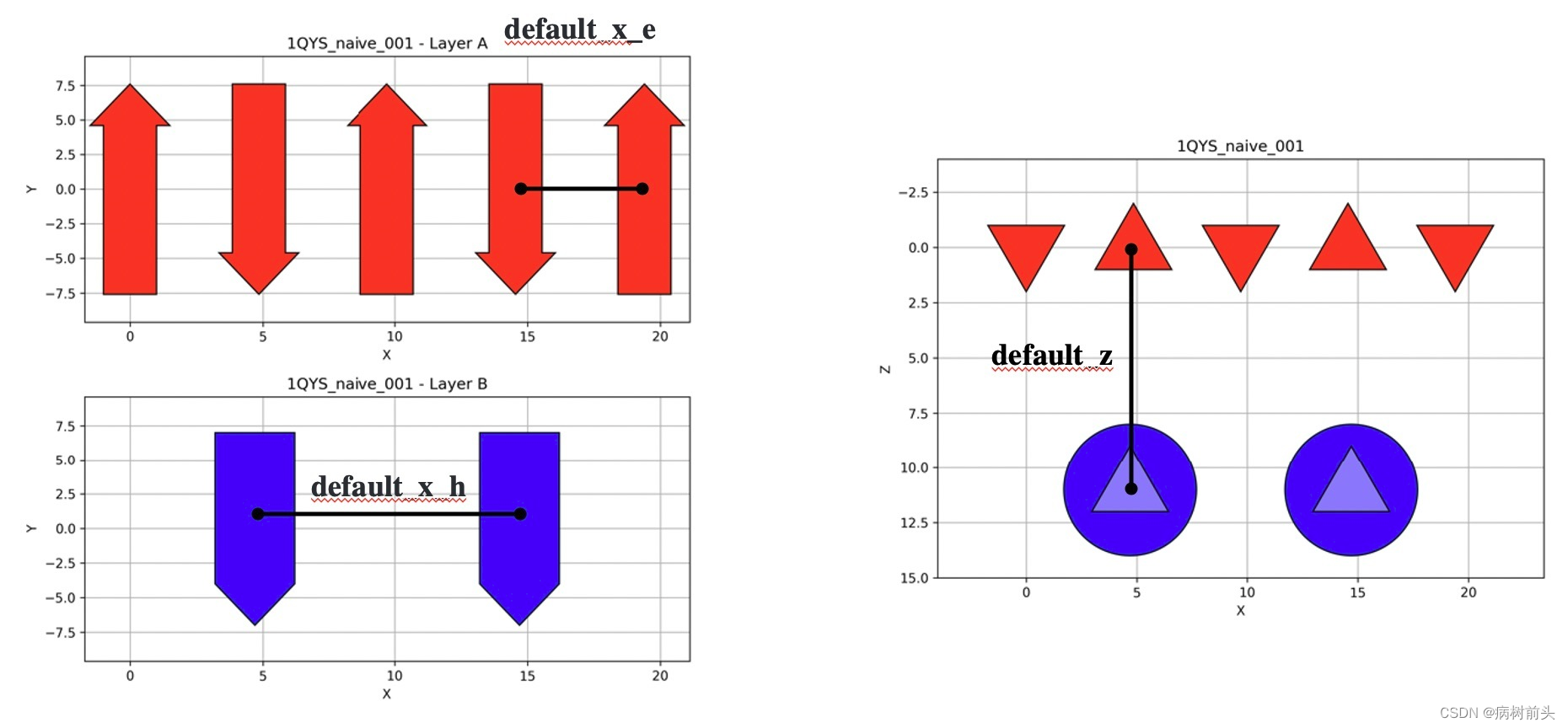 default_z: 两层layer之间的距离(默认: 11埃)
default_z: 两层layer之间的距离(默认: 11埃)
default_x_h: 同一层中螺旋结构之间的距离(默认: 11埃)
default_x_e: 同一层中β折叠片之间的距离(默认:5埃)
除了全局的描述以外,还可以更加精细地定义二级结构之间的连接方式:
link_dist: 默认两个二级结构末端之间的距离小于截断值,只有实际距离小于该值设定时,才考虑用Loop将其连起。
l_linkers: 定义连接两个二级结构的Loop长度,缺省时,将自动计算不同二级结构之间所需的最小Loop长度。
connectivity: 用户还可以定义二级结构之间的连接顺序。
除此以外,config中还必须定义Rosetta的基本路径信息:
name: 任务名
vall: Rosetta的vall数据库的绝对路径
rbin: Rosetta rosetta_scripts app的绝对路径
- Motifs属性
motifs是外源具有功能性的蛋白片段,通过motif属性我们可以从特定的PDB文件中指定该序列,并分离这个区域出来。
id: 定义字段的编号(字符标签,无实际意义)
pdbfile: 输入的pdb文件
chain: motif所在的pdb链ID
segments:
ini: pdb中motif片段的起始编号
end: pdb中motif片段的末端编号
id: 该二级结构的标签名称

具体实例中,id"2fx7"的中定义了1个motif,该名为"helix"的motif来源于2fx7.pdb的链P 671-686号氨基酸。如果需要添加多个motif,只需要在segment中继续添加字段即可。motif也可以来源于不同的链,此时应该另起一个id,来定义另外一条链的motif信息。
二、TopoBuilder的使用方法
1. TopoBuilder的使用方法
应当注意,越复杂的fold,loop之间的连接方式越有限。
以demo为例: 读者可尝试根据第一节内容的解释去理解其中的含义。
{
"config": {
"name": "4hb",
"vall": "/usr/local/rosetta_src_2019.47.61047_bundle/tools/fragment_tools/vall.jul19.2011.gz",
"rbin": "/usr/local/rosetta_src_2019.47.61047_bundle/main/source/bin/rosetta_scripts.mpi.macosclangrelease"
},
"layers" : [
[
{ "type" : "H",
"length" : 16,
"shift_y": 10,
"tilt_z":50
},
{ "type" : "H",
"length" : 16
}
],
[
{ "type" : "H",
"ref": "2fx7.helix"
},
{ "type" : "H",
"length" : 16
}
]
],
"motifs": [
{
"id": "2fx7",
"pdbfile": "2fx7.pdb",
"chain": "P",
"segments": [
{
"ini": 671,
"end": 686,
"id": "helix"
}
]
}
]
}


















 411
411











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









