










三叶青项目记录系列(二)
文章目录
前言
2024/1/21
主要做了对vcf原文件进行处理,然后筛选出位点
2024/7/22 更新:
有关于如何得到vcf原始文件,如有需要,请自行参考另一篇博客:重测序数据处理得到vcf文件
一、在Linux本地终端
因为老师给了我一个经过过滤后的SNP文件(没过滤的竟然有100多G),过滤后也有3.3个G,感觉也太大了,我还是在Linux终端用脚本跑吧
既然决定以后多多在linux中跑,那就干脆创建一个python的虚拟环境,用来管理这个项目 (推荐直接使用conda来管理项目,至于以下内容为很久之前记录的,可以选择忽视)
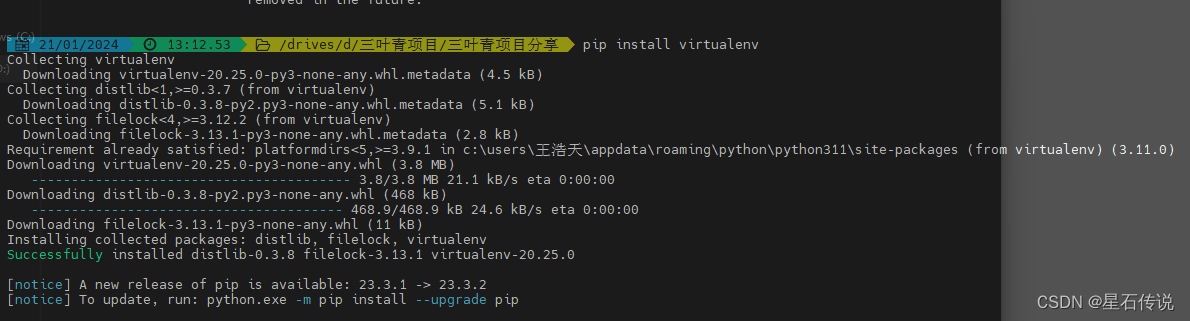
a. 安装虚拟环境工具(如果尚未安装):
pip install virtualenv
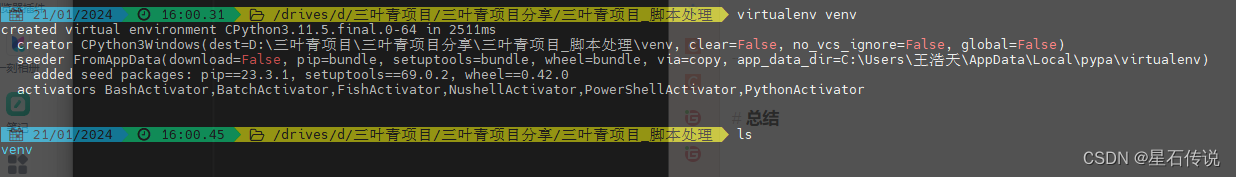
b. 在项目目录中创建一个新的虚拟环境:
cd /path/to/your/project
virtualenv venv
c. 激活虚拟环境:
激活后,你会注意到终端命令行前面有(venv)字样,表示你已进入虚拟环境。
source ./Scripts/activate
d. 完成项目工作后,可以停止虚拟环境:
deactivate

e. 在虚拟环境中安装依赖项:
以pandas包为例
pip install -i https://pypi.tuna.tsinghua.edu.cn/simple pandas
f. 运行脚本
python txt_transform_csv.py 'D:\三叶青项目\三叶青项目分享\139.SNP.gt.txt' 'D:\三叶青项目\三叶青项目分享\139.SNP.csv'
python vcf数据处理.py
二、远程连接服务器
突然发现在本地跑还是太慢了(根本不可能),不行,还是用服务器跑吧
用easyconnet实现连接VPN,下载地址:
https://pc.qq.com/detail/8/detail_26728.html
看一下我之前在服务器中创建的虚拟环境,然后激活
conda env list
conda activate python3
然后就在该环境中下载脚本运行所需依赖包
vcf文件处理
对139个样本的vcf文件进行处理:
#将前面的注释信息去掉,只保留矩阵数据
cat 139_文件名.vcf | tail -n +245 >xxx.vcf
#生成gt.txt文件(使用脚本vcf_keep_gt_only_1.py)
cat xxx.vcf | python vcf_keep_gt_only_1.py >xxx.gt.txt
#将矩阵中的所有的斜杠(/)替换为竖线(|)
sed -i 's/\//\|/g' xxx.gt.txt
#提取出vcf文件中的1、2、4、5行
awk '{print $1,$2,$4,$5}' xxx.vcf >site.txt
#用awk 和paste 把 ref和alt 列合并到gt.txt文件中
awk '{print $3,$4}' site.txt>site2.txt
paste site2.txt xxx.gt.txt >139.gt.txt
#然后使用脚本将txt文件转换为csv文件(方便后面直接得到表格结果)
python txt_transform_csv.py 139.gt.txt 139.gt.csv
#筛选出位点
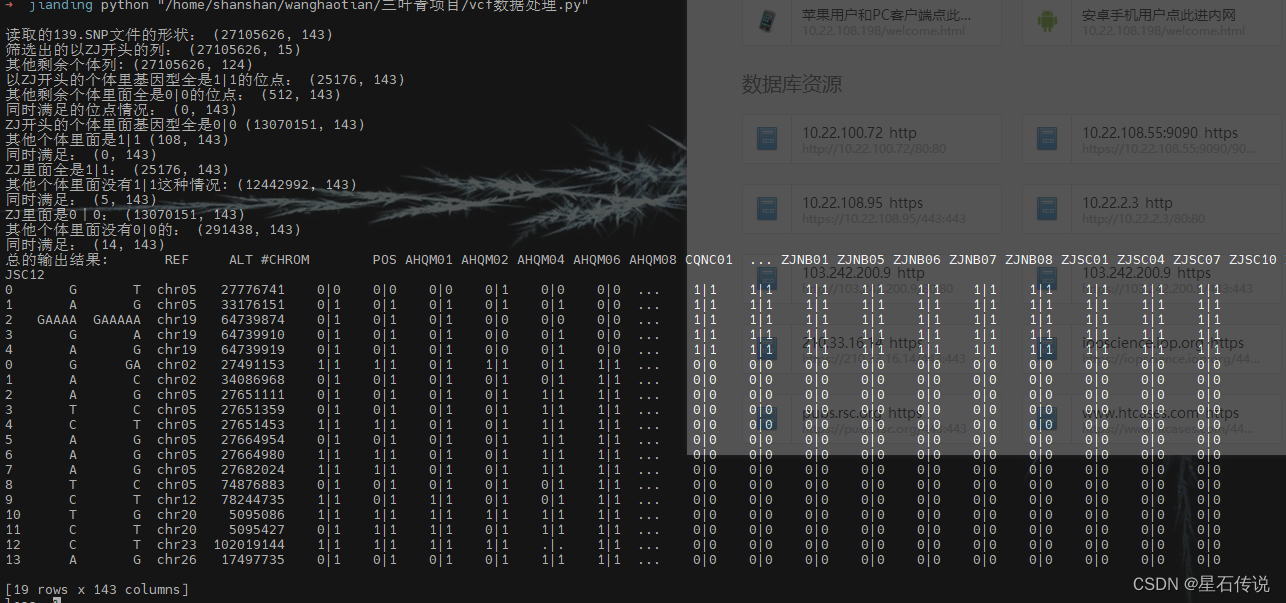
python vcf数据处理.py
三、代码补充
以下代码仅供参考,不太具有普适性,主要是针对当前任务进行
3.1. vcf_keep_gt_only_1.py
#!/usr/bin/env python3
##Usage: cat *.vcf|python vcf_keep_gt_only_1 > out_gt.txt
import sys
for line in sys.stdin:
if line[0:2] == "##":
next
elif line[0:2] =="#C":
a = line.strip().split("\t")
print(a[0]+"\t"+a[1]+"\t" + "\t".join(a[9:]))
else:
b = line.strip().split("\t")
GT_lst=b[9:]
GT_lst_n = [x.split(":")[0] for x in GT_lst]
GT_n = "\t".join(GT_lst_n)
print(b[0] + "\t" + b[1] + "\t" + GT_n )
3.2. txt_transform_csv.py
#!/usr/bin/env python3
import csv
def convert_txt_to_csv(txt_file, csv_file):
with open(txt_file, "r") as file:
txt_data = file.readlines()
data = []
for line in txt_data:
columns = line.strip().split("\t")
# 将第一列和第二列拆分为两个列
ref_alt = columns[0].split()
columns = ref_alt + columns[1:]
data.append(columns)
with open(csv_file, "w", newline="") as file:
writer = csv.writer(file)
writer.writerows(data)
print("转换完成!")
if __name__ == '__main__':
import argparse
parser = argparse.ArgumentParser(description='Convert a tab-separated text file to a comma-separated value file.')
parser.add_argument('input_file', metavar='input_file', type=str, help='The path to the input text file.')
parser.add_argument('output_file', metavar='output_file', type=str, help='The path to the output CSV file.')
args = parser.parse_args()
convert_txt_to_csv(args.input_file, args.output_file)
3.3. vcf数据处理.py
#!/usr/bin/env python3
import pandas as pd
# 读取数据文件
data = pd.read_csv('./139_wht.gt.csv', delimiter=',')
print('读取的139.SNP文件的形状:',data.shape)
# 筛选以"ZJ"开头的列
zj_individuals = data.filter(regex=r'^ZJ.*$', axis=1)
print('筛选出的以ZJ开头的列:',zj_individuals.shape)
#其它剩余个体列
other_individuals = data.drop(zj_individuals.columns, axis=1)
other_individuals = other_individuals.drop(data.columns[:4],axis=1)
print('其他剩余个体列:',other_individuals.shape)
# print(other_individuals.head(10))
# 先尝试找出在浙江也就是ZJ开头的个体里面基因型全是1|1 而在其他个体里面是0|0这种位点
zj_filtered01 = data[(zj_individuals == '1|1').all(axis=1)]
print('以ZJ开头的个体里基因型全是1|1的位点:',zj_filtered01.shape)
#print(zj_filtered01.head(10))
other_filtered01 = data[(other_individuals == '0|0').all(axis=1)]
print('其他剩余个体里面全是0|0的位点:',other_filtered01.shape)
#print(other_filtered01.head(10))
# 找到交集
filtered_data01 = pd.merge(zj_filtered01, other_filtered01, how='inner')
print('同时满足的位点情况:',filtered_data01.shape) #没有交集
# 或者找出在浙江也就是ZJ开头的个体里面基因型全是0|0 而在其他个体里面是1|1这种位点。
zj_filtered02 = data[(zj_individuals == '0|0').all(axis=1)]
print('ZJ开头的个体里面基因型全是0|0',zj_filtered02.shape)
other_filtered02 = data[(other_individuals == '1|1').all(axis=1)]
print('其他个体里面是1|1',other_filtered02.shape)
# 找到交集
filtered_data02 = pd.merge(zj_filtered02, other_filtered02, how='inner')
print('同时满足:',filtered_data02.shape)
# 可以找一下在ZJ里面全是1|1,在其他个体里面没有1|1这种情况
zj_filtered03 = data[(zj_individuals == '1|1').all(axis=1)]
print('ZJ里面全是1|1:',zj_filtered03.shape)
# print(zj_filtered03.head(10))
other_filtered03 = data[(other_individuals != '1|1').all(axis=1)]
print('其他个体里面没有1|1这种情况:',other_filtered03.shape)
# 找到交集
filtered_data03 = pd.merge(zj_filtered03, other_filtered03, how='inner')
print('同时满足:',filtered_data03.shape)
# print(filtered_data03)
# 还可以找一下在ZJ里面是0|0,在其他个体里面没有0|0的
zj_filtered04 = data[(zj_individuals == '0|0').all(axis=1)]
print('ZJ里面是0|0:',zj_filtered04.shape)
other_filtered04 = data[(other_individuals != '0|0').all(axis=1)]
print('其他个体里面没有0|0的:',other_filtered04.shape)
# 找到交集
filtered_data04 = pd.merge(zj_filtered04, other_filtered04, how='inner')
print('同时满足:',filtered_data04.shape)
# print(filtered_data04)
filtered_data = pd.concat([filtered_data01, filtered_data02,filtered_data03, filtered_data04])
# 输出结果
print('总的输出结果:',filtered_data)
# 保存结果
filtered_data.to_csv('filtered_data.csv', index=False)


















 2664
2664

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











