






 GWAS分析涉及复杂步骤,包括材料收集、取样、序列比对和变异鉴定。筛选候选位点可通过设定不同模型和阈值,如线性回归、MLM等,结合多种软件进行联合分析。精细定位如KASP技术可缩小定位区间。通过功能注释、多组学分析和选择清除验证,增强候选基因的可信度。
GWAS分析涉及复杂步骤,包括材料收集、取样、序列比对和变异鉴定。筛选候选位点可通过设定不同模型和阈值,如线性回归、MLM等,结合多种软件进行联合分析。精细定位如KASP技术可缩小定位区间。通过功能注释、多组学分析和选择清除验证,增强候选基因的可信度。

GWAS(全基因组关联分析)虽然是当前性状与基因型关联的有效方法,但是需要经过一连串的实验准备与数据分析。从前期的材料收集,到取样建库,序列比对,变异鉴定等重重步骤,最终通过GWAS在数百万个SNP/INDEL中鉴定出的位点如何确定就是我们想要的候选位点或者基因,下游的分析如何开展,主要有以下几种分析思路:
一:根据不同的模型与阈值进行显著位点的鉴定
如同前文,我们对于不同筛选阈值的讨论可以看出(什么是Bonferroni校正?| 群体遗传专题),阈值的确定对于鉴定出的位点数量有着直接的关系,根据阈值=0.05/有效变异数,那么对于有效的SNP/INDEL的变异数量的估计也是需要纳入考虑的[1]。另外,随着不同的关联分析模型的提出,常用的全基因组分析软件有数十个之多。早期,尽管线性或逻辑回归通常用于全基因组关联分析,但是其很难将包括群体结构和亲缘关系等因素纳入分析,随着例如MLM(mixed linear models)的提出,可以无需将这些因素分开,直接纳入分析,不幸的是,但是相对于简单的模型,MLM在计算上是十分耗时的。之后便有这许多生物信息工作者提出了许多的优化方法,如:FaST-LMM, GRAMMAR,MrMLM,Emmax等多种解决方案的提出。这些不同的模型在不同的数据基础上进行分析,所以最终得到的关联信号的位置以及信号的强弱均存在差异。所以,基于对于不同的软件对于SNP/INDEL的检出存在差异,在对表型的准确度比较确信的基础上,可以尝试使用多个软件进行联合分析,或许可以鉴定出不同的关联信号。
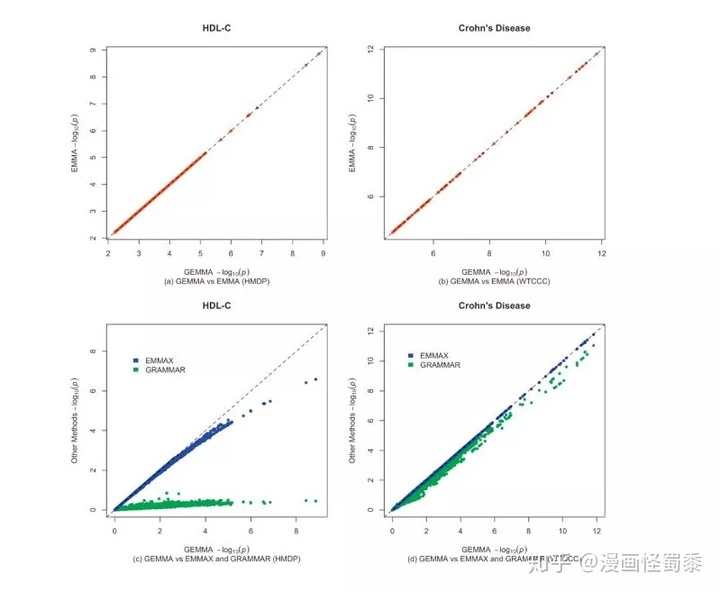
不同软件方法方法在相同的数据集上的表现略有差异在这种方法得到GWAS的候选位点之后,我们可以根据位点的位置获得位点附近区域的基因(基于LD block等方式估算的区间大小),并且对这些基因进行功能注释。根据功能注释,我们可以大致判断哪些基因是可能与我们所关注的性状相关的,并进入后续验证。如果有关联程度较为显著的非同义位点,可以尝试对该非同义位点进行功能验证。
二:KASP精细定位
通过设计引物来进一步缩小定位区段,精细定位对于挖掘候选基因也是十分有效的手段。基于区段内的变异信息,进行KASP(Kompetitive Allele Specific PCR) 高通量SNP分型技术可以帮助研究人员快速的在群体中对于SNP进行鉴定。由于SNP具有分布密度高、遗传稳定性好、二等位基因型等特点,可以解决实验人员面临区段内无常规引物可用的情况。KASP技术十分适合在大规模的群体中完成高通量的检测与更加精准的缩小定位区间。所以在GWAS得到结果后,可以在区间周围选择群体内具有多态性的位点,扩大群体进行基因分型,在更大的群体当中对关联信号的真实性进行检验。
三:多种组学与策略齐下
对于关联区域,如果呈现变异簇的形式,结合LD block分析确定出区段之后。我们可以检查区域内变异的注释信息,对于造成错义与转录终止的变异往往是我们较为关注的。如果变异较多,选择具有单倍型差异的样本进行RNA-seq来进行多组学联合分析也是一种有效的方式。结合转录组的差异分析,区域内差异表达的基因很有可能就是我们所要寻找的目标分析。当然,针对特定关注的性状,预先构建一个分离群体来选取极端性状进行混池测序来进行BSA定位也是一个很好的验证手段。除此之外,如果在初期选取了合适的群体,可以在进行GWAS分析的同时进行选择清除分析的操作,在目的性状具有显著差异的群体之间的选择清除信号如果与GWAS的关联信号相重合,说明GWAS的结果具有很高的可信度,如下图中桃重测序的分析结果,不同性状的GWAS关联信号分别能够与选择清除的信号峰对应上,说明了GWAS信号的可信性。

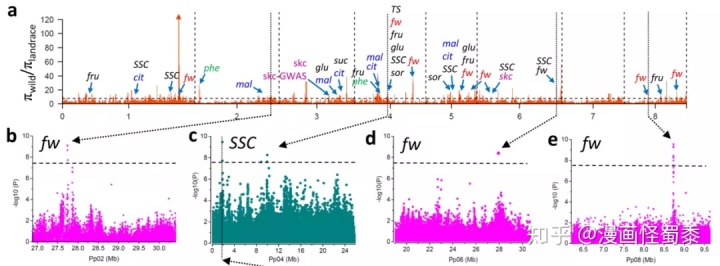
桃重测序不同性状GWAS信号与选择清除对应信号的关系
参考文献Browning, B. L. & Browning, S. R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126 (2016).Li Y, Cao K, Zhu G, et al. Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome biology, 2019, 20(1): 36.

















 3009
3009

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









