







oncoPredict
之前已经详细介绍过pRRophetic包预测药物敏感性了,但是这个包太古老了,我估计很多人会困在安装这一步,毕竟对于新手来说最难的就是R包安装了。
今天介绍下oncoPredict,这个包是pRRophetic的升级版,使用方法和原理一模一样,只是换了以下训练数据而已,也就是默认适用的数据库不一样了,其他都是一样的。除此之外还增加了几个新的函数。
主要功能是预测药物反应和药物-基因关联,github的描述:An R package for drug response prediction and drug-gene association prediction. The prepared GDSC and CTRP matrices for the calcPhenotype() are located in the oncoPredict OSF.
oncoPredict包的作者说他们会持续更新这个包,你们信吗?

CRAN和github都显示上次更新是2年前了~
安装
可以直接从cran安装了:
install.packages("oncoPredict")
但是安装后不能直接使用,需要下载这个包配套的训练数据,不过理论上你自己准备训练数据也是可以的~等我有时间尝试一下。
配套训练数据下载地址:https://osf.io/c6tfx/,一共是600多M大小。
library(oncoPredict)
##
##
## Attaching package: 'oncoPredict'
## The following objects are masked from 'package:pRRophetic':
##
## calcPhenotype, homogenizeData, summarizeGenesByMean
下载下来的训练数据就是以下几个,主要是GDSC1和GDSC2,以及CTRP的数据:

CTRP提供了RPKM和TPM两种格式的表达矩阵,GDSC则是芯片格式的。
简单看下数据是什么样子的:
GDSC2_Expr <- readRDS(file="../000files/DataFiles/Training Data/GDSC2_Expr (RMA Normalized and Log Transformed).rds")
GDSC2_Res <- readRDS(file = "../000files/DataFiles/Training Data/GDSC2_Res.rds")
dim(GDSC2_Expr) #17419,805
## [1] 17419 805
dim(GDSC2_Res) #805,198
## [1] 805 198
GDSC2_Expr[1:4,1:4]
## COSMIC_906826 COSMIC_687983 COSMIC_910927 COSMIC_1240138
## TSPAN6 7.632023 7.548671 8.712338 7.797142
## TNMD 2.964585 2.777716 2.643508 2.817923
## DPM1 10.379553 11.807341 9.880733 9.883471
## SCYL3 3.614794 4.066887 3.956230 4.063701
GDSC2_Res[1:4,1:4]
## Camptothecin_1003 Vinblastine_1004 Cisplatin_1005
## COSMIC_906826 -1.152528 -1.566172 7.017559
## COSMIC_687983 -1.263248 -4.292974 3.286848
## COSMIC_910927 -3.521093 -5.008028 2.492692
## COSMIC_1240138 1.976381 NA NA
## Cytarabine_1006
## COSMIC_906826 2.917980
## COSMIC_687983 2.790819
## COSMIC_910927 -1.082517
## COSMIC_1240138 NA
GDSC2_Expr是标准的表达矩阵格式,行是基因,列是细胞系,一共17419个基因,805个细胞系;
GDSC2_Res是每个细胞系对每个药物的IC50值,行是细胞系,列是药物,一共805种细胞系,198个药物。
再看下CTRP的数据,以TPM为例:
CTRP2_Expr <- readRDS(file="../000files/DataFiles/Training Data/CTRP2_Expr (TPM, not log transformed).rds")
CTRP2_Res <- readRDS(file = "../000files/DataFiles/Training Data/CTRP2_Res.rds")
dim(CTRP2_Expr)
## [1] 51847 829
dim(CTRP2_Res)
## [1] 829 545
CTRP2_Expr[1:4,1:4]
## CVCL_1045 CVCL_1046 CVCL_7937 CVCL_7935
## DDX11L1 0.0000000 0.10511906 0.0000000 0.1444173
## WASH7P 34.3887920 28.39068559 14.8423404 14.5556952
## MIR1302-11 0.1167793 0.02903022 0.4054605 0.5185439
## FAM138A 0.0000000 0.02432715 0.5362218 0.3674906
CTRP2_Res[1:4,1:4]
## CIL55 BRD4132 BRD6340 BRD9876
## CVCL_1045 14.504 14.819 14.101 14.657
## CVCL_1046 14.982 12.110 14.529 14.702
## CVCL_7937 14.388 12.091 14.742 13.230
## CVCL_7935 14.557 NA 14.546 13.258
和GDSC的数据格式一模一样的,也是表达矩阵和药敏信息,一共51847个基因,应该是包括mRNA和非编码RNA的,829个细胞系,545种药物。
calcPhenotype
这个包主要就是3个函数,最重要的一个就是calcPhenotype了。pRRophetic也有calcPhenotype函数,其实都是一样的用法,各个参数我们在上一篇中也介绍过了,这里就不多说了。
我们使用TCGA-BLCA的lncRNA的表达矩阵进行演示,因为CTPR的训练数据是转录组,而且包含了非编码RNA,所以是可以用来预测lncRNA的药物敏感性的。
下面这个示例数据我放在了粉丝QQ群,需要的加群下载即可。
首先加载lncRNA的表达矩阵:
load(file = "../000files/testExpr_BLCA.rdata")
这个示例表达矩阵一共12162个lncRNA,414个样本,并且被分为高风险组和低风险组,表达矩阵已经过了log2转换:
table(sample_group)
## sample_group
## high low
## 207 207
dim(testExpr)
## [1] 12162 414
testExpr[1:4,1:4]
## TCGA-ZF-A9R5-01A-12R-A42T-07 TCGA-E7-A7PW-01A-11R-A352-07
## AL627309.1 0.01705863 0.00000000
## AL627309.2 0.09572744 0.00000000
## AL627309.5 0.08029061 0.01252150
## AC114498.1 0.00000000 0.06007669
## TCGA-4Z-AA7N-01A-11R-A39I-07 TCGA-DK-A2I1-01A-11R-A180-07
## AL627309.1 0.00000000 0.000000000
## AL627309.2 0.00000000 0.000000000
## AL627309.5 0.04722951 0.004103184
## AC114498.1 0.00000000 0.000000000
下面就可以一次计算所有545种药物的敏感性了,速度很慢,而且结果只能保存到当前工作目录下的calcPhenotype_Output文件夹中。
calcPhenotype(trainingExprData = CTRP2_Expr,
trainingPtype = CTRP2_Res,
testExprData = as.matrix(testExpr),#需要matrix
batchCorrect = 'eb',
#IC50是对数转换的,所以表达矩阵也用对数转换过的
powerTransformPhenotype = F,
minNumSamples = 20,
printOutput = T,
removeLowVaryingGenes = 0.2,
removeLowVaringGenesFrom = "homogenizeData"
)
结果就一个文件,就是每个样本对每一个药物的IC50值,读取进来看看:
res <- read.csv("./calcPhenotype_Output/DrugPredictions.csv")
dim(res)
## [1] 414 546
res[1:4,1:4]
## X CIL55 BRD4132 BRD6340
## 1 TCGA-ZF-A9R5-01A-12R-A42T-07 14.10501 13.45597 14.30078
## 2 TCGA-E7-A7PW-01A-11R-A352-07 13.81229 13.19243 14.10742
## 3 TCGA-4Z-AA7N-01A-11R-A39I-07 14.02137 13.02397 14.27905
## 4 TCGA-DK-A2I1-01A-11R-A180-07 14.48769 13.54434 14.41776
这样就得到了414个样本对每种药物的IC50值。
有了这个结果,我们就可以取出感兴趣的药物,可视化IC50值在不同组间的差异,和之前介绍的一模一样,我们这里随便取前10个药物:
library(tidyr)
library(dplyr)
library(ggplot2)
library(ggpubr)
library(ggsci)
res %>%
select(1:11) %>%
bind_cols(sample_group = sample_group) %>%
pivot_longer(2:11,names_to = "drugs",values_to = "ic50") %>%
ggplot(., aes(sample_group,ic50))+
geom_boxplot(aes(fill=sample_group))+
scale_fill_jama()+
theme(axis.text.x = element_text(angle = 45,hjust = 1),
axis.title.x = element_blank(),
legend.position = "none")+
facet_wrap(vars(drugs),scales = "free_y",nrow = 2)+
stat_compare_means()
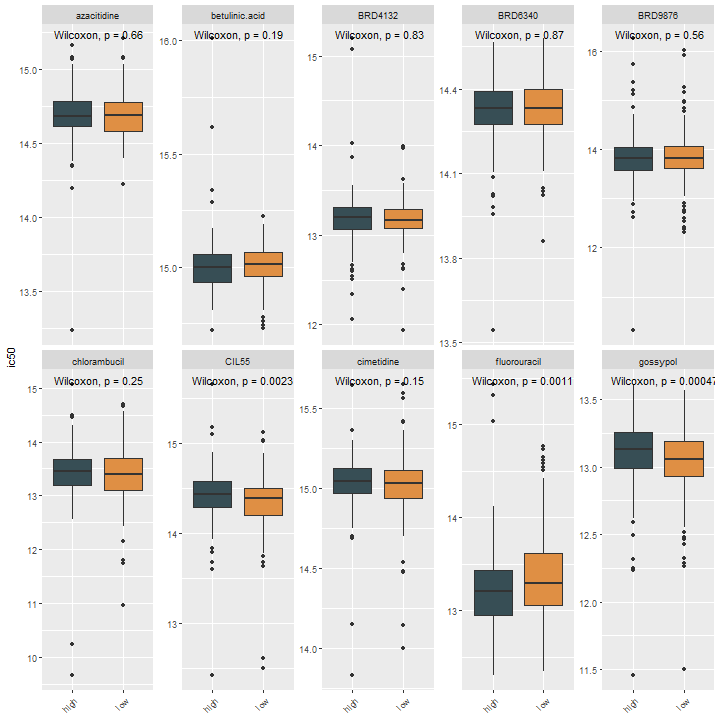
也是一样的easy。
除此之外,oncoPredict包还有其他几个函数,我们简单介绍一下。
IDWAS
IDWAS 方法是一种扩展的药物响应值填补方法,可以方便地在人群中进行生物标志物的鉴定。IDWAS 在概念和实施上与GWAS相似,即通过使用R中的线性模型确定填补的药物响应值与体细胞突变或拷贝数变异之间的关联,以估计药物-基因相互作用并识别药物响应的生物标志物。利用临床数据集的大样本量,这种方法可以发现细胞系数据集中没有的新关系。此外,由于使用了患者的基因组数据,所找到的任何生物标志物都与该患者群体相关。
简单来说就是根据药敏信息和表型信息识别药物的靶点。表型信息可以是突变数据或者拷贝数变异数据。
CNV
我们这里先试试拷贝数变异,直接使用TCGA-BLCA的拷贝数变异数据,直接使用easyTCGA,1行代码搞定拷贝数变异数据的下载:
rm(list = ls())
library(oncoPredict)
library(easyTCGA)
getcnv("TCGA-BLCA")
下载完成后直接加载数据即可:
load(file = "G:/easyTCGA_test/output_cnv/TCGA-BLCA_CNV.rdata")
blca_cnv <- data
head(blca_cnv)
## # A tibble: 6 × 7
## GDC_Aliquot Chromosome Start End Num_Probes Segment_Mean Sample
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <chr>
## 1 64caa1a2-b01f-404c-ba… 1 3.30e6 1.57e8 71640 0.0136 TCGA-…
## 2 64caa1a2-b01f-404c-ba… 1 1.57e8 1.57e8 2 -1.66 TCGA-…
## 3 64caa1a2-b01f-404c-ba… 1 1.57e8 1.86e8 18893 0.0124 TCGA-…
## 4 64caa1a2-b01f-404c-ba… 1 1.86e8 1.86e8 2 -1.79 TCGA-…
## 5 64caa1a2-b01f-404c-ba… 1 1.86e8 2.48e8 39130 0.0141 TCGA-…
## 6 64caa1a2-b01f-404c-ba… 2 4.81e5 2.42e8 132068 0.0091 TCGA-…
然后使用map_cnv转换一下格式。
The mapping is accomplished by intersecting the gene with the overlapping CNV level. If the gene isn’t fully #captured by the CNV, an NA will be assigned.
map_cnv(blca_cnv)
403 genes were dropped because they have exons located on both strands of the
same reference sequence or on more than one reference sequence, so cannot be
represented by a single genomic range.
Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
object, or use suppressMessages() to suppress this message.
生成的map.RData文件会自动保存在当前工作目录下,加载进来:
load(file = "./map.RData")
data<-as.data.frame(t(theCnvQuantVecList_mat))
接下来是准备药敏数据,这个数据我们可以直接使用上面的calcPhenotype得到的结果:
drug_prediction<-as.data.frame(read.csv('./calcPhenotype_Output/DrugPredictions.csv', header=TRUE, row.names=1))
dim(drug_prediction)
## [1] 414 545
drug_prediction[1:4,1:4]
## CIL55 BRD4132 BRD6340 BRD9876
## TCGA-ZF-A9R5-01A-12R-A42T-07 14.10501 13.45597 14.30078 14.05845
## TCGA-E7-A7PW-01A-11R-A352-07 13.81229 13.19243 14.10742 13.79403
## TCGA-4Z-AA7N-01A-11R-A39I-07 14.02137 13.02397 14.27905 12.32300
## TCGA-DK-A2I1-01A-11R-A180-07 14.48769 13.54434 14.41776 12.53436
然后就可以使用idwas计算了:
idwas(drug_prediction=drug_prediction, data=data, n=10, cnv=T)
结果文件会自动保存在当前工作目录中。
cnvPvalue <- read.csv("CnvTestOutput_pVals.csv",row.names = 1)
dim(cnvPvalue)
## [1] 545 402
cnvPvalue[15:20,1:4]
## SNORD123 MIR875 MKRN2OS LOC100130075
## teniposide 0.157018358 0.8511244 0.08231962 0.05243646
## sildenafil 0.057896196 0.1650301 0.11829531 0.16373477
## simvastatin 0.003648419 0.1894510 0.16468764 0.24833938
## parbendazole 0.309342093 0.6758557 0.09965932 0.74859683
## procarbazine 0.784280696 0.3716664 0.11423190 0.42647709
## curcumin 0.297120449 0.6824545 0.73916619 0.23619591
行是药物,列是找出来的靶点,中间是P值,应该是P值小于0.05说明有意义吧,详细情况大家自己阅读文献了解。
还有一个beta-value矩阵,一样的格式,就不展示了。
snv
如果你使用的表型数据是突变数据,那就更简单了,这里还是以TCGA-BLCA的突变数据为例。
直接使用easyTCGA1行代码下载突变数据:
library(easyTCGA)
getsnvmaf("TCGA-BLCA")
药敏数据还是和上面一样的,把突变数据直接加载进来就可以进行计算了。
因为这里使用的是突变数据不是拷贝数变异,所以需要更改参数cnv=F,其他完全一样:
load(file = "G:/easyTCGA_test/output_snv/TCGA-BLCA_maf.rdata")
idwas(drug_prediction=drug_prediction, data=data, n=10, cnv=F)
结果文件也是自动保存在当前工作目录中。
GLDS
作者在原文中对GLDS的解释:
我们之前就“GLDS 现象”及其对生物标志物鉴定的影响进行了报道。GLDS是指在一个人群(细胞系或患者)中,无论接受何种治疗,个体通常可以表现出更敏感或更耐药的趋势。正如原始论文所示,这一现象与多重耐药(MDR)有关,但并不完全相同。对这一变量进行校正可以更具体地确定与特定药物相关的生物标志物。
也就是说这个函数可以根据你提供的表型数据和药敏数据,推测出与药物最相关的靶点,感觉和上面的函数作用差不多。
表型数据也可以是拷贝数变异或者突变数据。
下面是一个示例。
首先加载药敏数据,我这里用的是示例数据和示例代码。
rm(list = ls())
trainingPtype = readRDS(file = "../000files/DataFiles/Training Data/GDSC2_Res.rds")
class(trainingPtype)
## [1] "matrix" "array"
dim(trainingPtype)
## [1] 805 198
trainingPtype[1:4,1:4]
## Camptothecin_1003 Vinblastine_1004 Cisplatin_1005
## COSMIC_906826 -1.152528 -1.566172 7.017559
## COSMIC_687983 -1.263248 -4.292974 3.286848
## COSMIC_910927 -3.521093 -5.008028 2.492692
## COSMIC_1240138 1.976381 NA NA
## Cytarabine_1006
## COSMIC_906826 2.917980
## COSMIC_687983 2.790819
## COSMIC_910927 -1.082517
## COSMIC_1240138 NA
行是细胞系,列是药物,也是一个表达矩阵的形式。
GLDS不能有缺失值,所以先进行缺失值插补,使用自带的completeMatrix函数,默认进行50次迭代,巨慢!!所以我设置成3了。
# 缺失值插补
completeMatrix(trainingPtype, nPerms = 3)
结果文件complete_matrix_output_GDSCv2.txt会自动保存在当前工作中.
# 读取插补后的药敏数据
cm<-read.table('complete_matrix_output.txt', header=TRUE, row.names=1)
dim(cm)
## [1] 805 198
cm[1:4,1:4]
## Camptothecin_1003 Vinblastine_1004 Cisplatin_1005
## COSMIC_906826 -1.152528 -1.566172 7.017559
## COSMIC_687983 -1.263248 -4.292974 3.286848
## COSMIC_910927 -3.521093 -5.008028 2.492692
## COSMIC_1240138 1.976381 -3.957938 3.354192
## Cytarabine_1006
## COSMIC_906826 2.917980
## COSMIC_687983 2.790819
## COSMIC_910927 -1.082517
## COSMIC_1240138 1.697800
为了进行计算,我们把细胞系和药物的名字都改一下,主要是去掉细胞系的前缀和药物的后缀。
读取细胞系的信息,里面有细胞系名字的详细信息:
cellLineDetails<-readxl::read_xlsx('../000files/DataFiles/GLDS/GDSCv2/Cell_Lines_Details.xlsx')
dim(cellLineDetails)
## [1] 1002 13
cellLineDetails[1:4,1:4]
## # A tibble: 4 × 4
## `Sample Name` `COSMIC identifier` `Whole Exome Sequencing (WES)`
## <chr> <dbl> <chr>
## 1 A253 906794 Y
## 2 BB30-HNC 753531 Y
## 3 BB49-HNC 753532 Y
## 4 BHY 753535 Y
## # ℹ 1 more variable: `Copy Number Alterations (CNA)` <chr>
替换药敏数据的行名,也就是细胞系名字:
newRows <- substring(rownames(cm),8) #Remove 'COSMIC'...keep the numbers after COSMIC.
indices<-match(as.numeric(newRows), as.vector(unlist(cellLineDetails[,2]))) #Refer to the cell line details file to make this replacement.
newNames<-as.vector(unlist(cellLineDetails[,1]))[indices] #Reports the corresponding cell line names
rownames(cm)<-newNames
dim(cm)
## [1] 805 198
cm[1:4,1:4]
## Camptothecin_1003 Vinblastine_1004 Cisplatin_1005 Cytarabine_1006
## CAL-120 -1.152528 -1.566172 7.017559 2.917980
## DMS-114 -1.263248 -4.292974 3.286848 2.790819
## CAL-51 -3.521093 -5.008028 2.492692 -1.082517
## H2869 1.976381 -3.957938 3.354192 1.697800
根据作者提供的信息,把药物名字也改一下:
fix <- readxl::read_xlsx('../000files/DataFiles/GLDS/GDSCv2/gdscv2_drugs.xlsx')
## New names:
## • `` -> `...2`
fix<-as.vector(unlist(fix[,1]))
head(fix)
## [1] "Camptothecin" "Vinblastine" "Cisplatin" "Cytarabine" "Docetaxel"
## [6] "Gefitinib"
colnames(cm)<-as.vector(fix)
drugMat<-as.matrix(cm) #Finally, set this object as the drugMat parameter.
dim(drugMat) #805 samples vs. 198 drugs
## [1] 805 198
drugMat[1:4,1:4]
## Camptothecin Vinblastine Cisplatin Cytarabine
## CAL-120 -1.152528 -1.566172 7.017559 2.917980
## DMS-114 -1.263248 -4.292974 3.286848 2.790819
## CAL-51 -3.521093 -5.008028 2.492692 -1.082517
## H2869 1.976381 -3.957938 3.354192 1.697800
到这里这个药敏数据终于准备好了!
下面要准备marker矩阵,也就是突变或者拷贝数变异数据。这里也是用的示例数据,是一个泛癌的数据,同时包含CNV和突变信息:
mutationMat<-read.csv('../000files/DataFiles/GLDS/GDSCv2/GDSC2_Pan_Both.csv')
mutationMat<-mutationMat[,c(1,6,7)] #Index to these 3 columns of interest.
colnames(mutationMat) #"cell_line_name" "genetic_feature" "is_mutated"
## [1] "cell_line_name" "genetic_feature" "is_mutated"
head(mutationMat)
## cell_line_name genetic_feature is_mutated
## 1 CAL-29 CDC27_mut 0
## 2 CAL-29 CDC73_mut 0
## 3 CAL-29 CDH1_mut 0
## 4 CAL-29 CDK12_mut 0
## 5 CAL-29 CDKN1A_mut 0
## 6 CAL-29 CDKN1B_mut 0
上面这个文件中有一些重复的cell_line_name和genetic_feature对,先去掉重复的:
vec<-c()
for (i in 1:nrow(mutationMat)){
vec[i]<-paste(mutationMat[i,1],mutationMat[i,2], sep=' ')
}
nonDupIndices<-match(unique(vec), vec)
mutationMat2<-mutationMat[nonDupIndices,]
dim(mutationMat2)
## [1] 584051 3
head(mutationMat2)
## cell_line_name genetic_feature is_mutated
## 1 CAL-29 CDC27_mut 0
## 2 CAL-29 CDC73_mut 0
## 3 CAL-29 CDH1_mut 0
## 4 CAL-29 CDK12_mut 0
## 5 CAL-29 CDKN1A_mut 0
## 6 CAL-29 CDKN1B_mut 0
把空值去掉,再变为宽数据,使得行名是细胞系名字:
library(tidyverse)
good<-(mutationMat2[,2]) != ""
mutationMat3<-mutationMat2[good,]
mutationMat4<-mutationMat3 %>%
pivot_wider(names_from=genetic_feature,
values_from=is_mutated)
rownames(mutationMat4)<-as.vector(unlist(mutationMat4[,1])) #Make cell lines the rownames...right now they are column 1.
cols<-rownames(mutationMat4)
mutationMat4<-as.matrix(t(mutationMat4[,-1]))
dim(mutationMat4)
## [1] 1315 1389
mutationMat4[1:4,1:4]
## [,1] [,2] [,3] [,4]
## CDC27_mut "0" "0" "0" NA
## CDC73_mut "0" "0" "0" NA
## CDH1_mut "0" "0" "0" NA
## CDK12_mut "0" "0" "0" NA
把字符型变为数值型,把NA也变成0:
#Make sure the matrix is numeric.
mutationMat<-mutationMat4
mutationMat4<-apply(mutationMat4, 2, as.numeric)
rownames(mutationMat4)<-rownames(mutationMat)
markerMat<-mutationMat4
markerMat[1:4,1:4]
## [,1] [,2] [,3] [,4]
## CDC27_mut 0 0 0 NA
## CDC73_mut 0 0 0 NA
## CDH1_mut 0 0 0 NA
## CDK12_mut 0 0 0 NA
# replace all non-finite values with 0
markerMat[!is.finite(markerMat)] <- 0
colnames(markerMat)<-cols
dim(markerMat) #1315 1389
## [1] 1315 1389
markerMat[1:4,1:4]
## CAL-29 CAL-33 697 CCNE1
## CDC27_mut 0 0 0 0
## CDC73_mut 0 0 0 0
## CDH1_mut 0 0 0 0
## CDK12_mut 0 0 0 0
#保存一下
#write.table(markerMat, file='markerMat.txt')
markerMat<-as.matrix(read.table('markerMat.txt', header=TRUE, row.names=1))
到这里marker矩阵也准备好了。
最后还需要一个drug relatedness file文件,前面改名字就是为了和这里的名字保持一致。
drugRelatedness: This file is GDSC’s updated drug relatedness file (obtained from bulk data download/all compounds screened/compounds-annotation).
drugRelatedness <- read.csv("../000files/DataFiles/GLDS/GDSCv2/screened_compunds_rel_8.2.csv")
drugRelatedness<-drugRelatedness[,c(3,6)]
colnames(drugRelatedness) #"DRUG_NAME" "TARGET_PATHWAY"
## [1] "DRUG_NAME" "TARGET_PATHWAY"
head(drugRelatedness)
## DRUG_NAME TARGET_PATHWAY
## 1 Erlotinib EGFR
## 2 Rapamycin PI3K/MTOR
## 3 Sunitinib RTK
## 4 PHA.665752 RTK
## 5 MG-132 Protein stability and degradation
## 6 Paclitaxel Mitosis
然后就可以运行glds了:
glds(drugMat,
drugRelatedness,
markerMat,
minMuts=5,
additionalCovariateMatrix=NULL,
threshold=0.7)
结果也是自动保存在当前工作目录中。
文献中的这张图应该就是根据上面的结果画出来的,大家自己研究下喽~
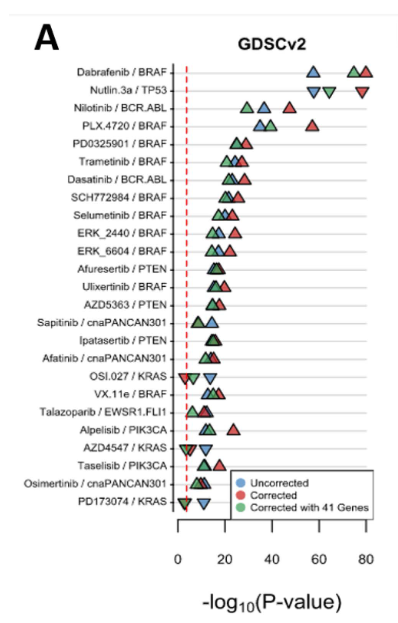
结束。
本文对后两个函数只是按照帮助文档运行了一遍,如果要详细了解各种细节,建议大家去阅读文献哈~

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









