







 本文详细介绍了在Windows电脑上安装Linux系统(Ubuntu22.04.4)的过程,涉及硬件信息配置、磁盘分区、制作系统盘、安装conda环境、Linux基础操作、常用软件下载、conda环境配置、sra数据下载与处理以及CellRanger的安装与运行。
本文详细介绍了在Windows电脑上安装Linux系统(Ubuntu22.04.4)的过程,涉及硬件信息配置、磁盘分区、制作系统盘、安装conda环境、Linux基础操作、常用软件下载、conda环境配置、sra数据下载与处理以及CellRanger的安装与运行。
文章目录
本文最后修改时间为2024/3/27
1 安装Linux系统
硬件信息
| 组件 | 具体硬件 |
|---|---|
| CPU | 13700KF 16C24T |
| 主板 | ROG STRIX Z690-A GAMING WIFI |
| 运行内存 | 英睿达 32GB*4 DDR5 4800Hz |
| GPU | Nvidia 4090 24G |
| 磁盘驱动器(作为Ubuntu系统盘) | 西数 SN580 2TB |
注:这台电脑之前已有安装Windows11专业版,对应的存储盘的参数在此省略
制作系统盘
64G的格式化后的空U盘,Ubuntu22.04.4,清华镜像也可以下载。从UltraISO软碟通中文官方网站 - 光盘映像文件制作/编辑/转换工具下载UltraISO试用版制作系统盘。
具体过程参考Windows+Ubuntu20.04双系统安装教程 - 知乎 (zhihu.com)
相信能看到这篇文章的人至少对英文操作系统不至于抓瞎,所以我推荐直接英文完成安装。一开始就改语言会把系统的真实路径一起给改了,可能会出奇怪的bug,装完后可以调语言的。
参考里创建分区有点问题:
- swap交换分区(虚拟内存),逻辑分区,我分了32GB,只要不超内存这个设置没问题
- boot分区 ,逻辑分区,默认ext4。 300M不够的,我装完后直接满了,然后总是弹窗提示我扩容,非常烦人,我索性改成2G了
- root分区,逻辑分区,我给了250G就可以,默认ext4,
- home分区,主分区,默认ext4,剩下的全部分给它了,1.6TB左右,用户的所有文件都在这里。
磁盘重新划分
如果你和我一样碰到了存储分配不当的问题,不拔U盘,继续走UEFI启动,在图形化界面那一步直接选择试用Ubuntu,可以参考Ubuntu扩展所有分区空间的完整教程_ubuntu扩展分区空间-CSDN博客
常用软件
-
Edge:我本人不太喜欢火狐和Chrome,下载常用软件,可以登文件传输助手
-
百度网盘:备份和互传文件用,缺点是网速十分感人,500M宽带下传1G文件都很痛苦。据说可以用SSH传文件,等有空再研究研究。
2 安装conda环境
熟悉windows下anaconda环境的朋友这一步应该是轻车熟路,按着教程做就完了,不作赘述,参考的这篇文章也是做上游的,本文要用到的包括sratoolkit、aspera、parrallel-fastq-dump
3 需要用到的一些Linux基础操作
常见命令
会用cd,ls就差不多了,其他的命令我也记不住,现用现查吧
新手慎用rm,在图形界面下手动删除
Ubuntu 20.04 LTS 系统自带的截图功能 - 肥斯大只仔 - 博客园 (cnblogs.com)
bash脚本和vim
vim操作在添加路径的时候要用到,简单的几个命令要记一下:Linux 之 Vim 命令使用(详细总结) - 知乎 (zhihu.com)
bash脚本和bat脚本就是换汤不换药,想提高效率可以读一下鸟叔的那本书,讲的很到位
4 下载sra数据
曾健明大佬的上游流程有点老了,可以参考着做,但是有很多坑:单细胞实战(一)数据下载 (qq.com),他写的脚本对于较小的下载没有问题,但是对于我这个样本就有点离谱了
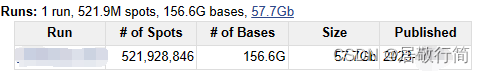
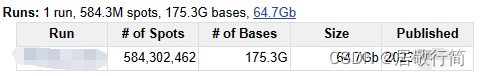
我是走ENA Browser下的lite文件,关于lite文件和sra文件的区别可以参考NCBI的SRA Lite和SRA Normalized数据有什么区别 - 组学大讲堂问答社区 (omicsclass.com)
workd="/home/williamhan/bioproject/MCAO20240324"
cd $workd/raw
# aspera密钥路径
openssh="/home/williamhan/miniconda3/envs/transcriptomics/etc/asperaweb_id_dsa.openssh"
# 参照https://zhuanlan.zhihu.com/p/667695791下载整理下file.lst, 名字不变
ascp -vQT -l 500m -P33001 -k 1 -i $openssh\
--host fasp.sra.ebi.ac.uk\
--user era-fasp\
--file-list file_sra.lst\
--mode recv\
./
-vverbose mode 唠叨模式,能让你实时知道程序在干啥,方便查错。有些作者的程序缺乏人性化,运行之后,只见光标闪,压根不知道运行到哪了
-T取消加密,否则有时候数据下载不了
-i提供私钥文件的地址,地址一般是~/miniconda3/etc/asperaweb_id_dsa.openssh
-l设置最大传输速度,一般200m到500m,如果不设置,反而速度会比较低,可能有个较低的默认值,不知道网速的上中国科学技术大学测速网站 (ustc.edu.cn)测一下看看
-k断点续传,一般设置为值1
-Q加入队列
-P提供SSH port,一般是33001
然后用parallel-fastq-dump基于lite文件把fastq.gz文件下载下来,关于我为啥不用曾老师教程里推荐的fastq-dump和官方推荐的fasterq-dump,可参见以下三个网址:
sratoolkit官方Github介绍:08. prefetch and fasterq dump · ncbi/sra-tools Wiki (github.com)
parallel-fastq-dump的Github页面:rvalieris/parallel-fastq-dump: parallel fastq-dump wrapper (github.com)
速度比较:fastq-dump、fasterq-dump和parallel-fastq-dump处理SRA文件的速度比较 - 简书 (jianshu.com)
workd="/home/williamhan/bioproject/MCAO20240324"
cd $workd/raw
cat SRR_Acc_List.txt |while read i
do
parallel-fastq-dump -t 12 -O ./${i} --split-files --gzip -s ${i}.lite
done
-t代表线程数
-O代表输出路径
--split-files代表拆分文件,一定要加!原因详见单细胞实战(二) cell ranger使用前注意事项 (qq.com)
--gzip压缩为.gz格式,这个一定要点,拿上面图中57.7Gb的文件为例,下载下来未经压缩的fastq文件加起来有250多G!
-ssra或lite文件地址
按照曾老师的教程,下载下来应该是3个文件,但是我始终是两个文件。我推测这个样本只上传了read文件。改名之后,只有R1和R2一样可以跑
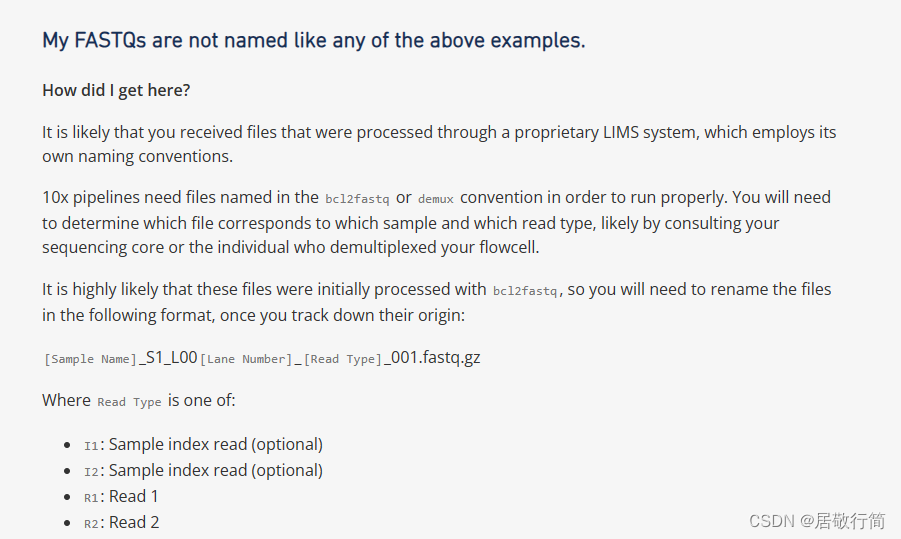
# 比如,将原来的SRR7692286_1.fastq.gz改成SRR7692286_S1_L001_I1_001.fastq.gz
# 依次类推,将原来_2的改成R1,将_3改成R2
# 但是有些文件只有两个的,说明不包含Index文件
# 改名方法是_1改为R1,_2改为R2
fq_dir=/home/williamhan/bioproject/MCAO20240324/raw
cd $fq_dir
cat $fq_dir/SRR_Acc_List.txt |while read i
do
# 三个文件改名
# mv ${i}_1*.gz ${i}_S1_L001_I1_001.fastq.gz;
# mv ${i}_2*.gz ${i}_S1_L001_R1_001.fastq.gz;
# mv ${i}_3*.gz ${i}_S1_L001_R2_001.fastq.gz;
# 两个文件改名:
mv ${i}/${i}.lite_1.fastq.gz ${i}_S1_L001_R1_001.fastq.gz;
mv ${i}/${i}.lite_2.fastq.gz ${i}_S1_L001_R2_001.fastq.gz;
done
5 cell ranger下载和运行
下载
参考链接:
官方下载链接:Download Cell Ranger - Official 10x Genomics Support
需要你填一下个人信息,填完就可以下载。

这三步是必须要做的
转录本我懒得从ENTREZ那里找了,直接从10x给的链接下了
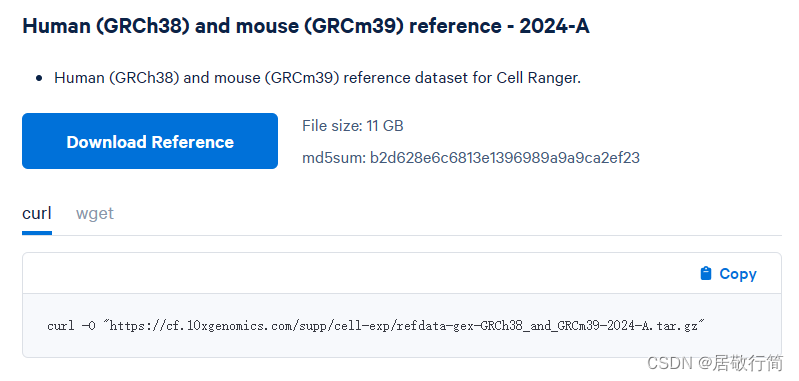
我装的版本的8.0.0,我和上面唯一的区别是我直接把cell ranger的路径添加为所有用户的全局变量了
sudo vim /etc/profile
运行
# cellranger定量
echo $(date "+%Y-%m-%d %H:%M:%S")
db=/home/williamhan/opt/refdata-gex-GRCh38_and_GRCm39-2024-A
ls $db
fq_dir=/home/williamhan/bioproject/MCAO20240324/raw
cat $fq_dir/SRR_Acc_List_PRJNA912889.txt |while read i
do cellranger count --id=$i --localcores=21 --create-bam false \
--transcriptome=$db --fastqs=${fq_dir}/${i} --sample=$i --nosecondary --expect-cells=10000
done
--id文件地址
--localcores线程数
--create-bam是否输出bam文件
--transcriptome转录本数据库,直接从官网下载就完了
12:40开始运行, 14:58结束
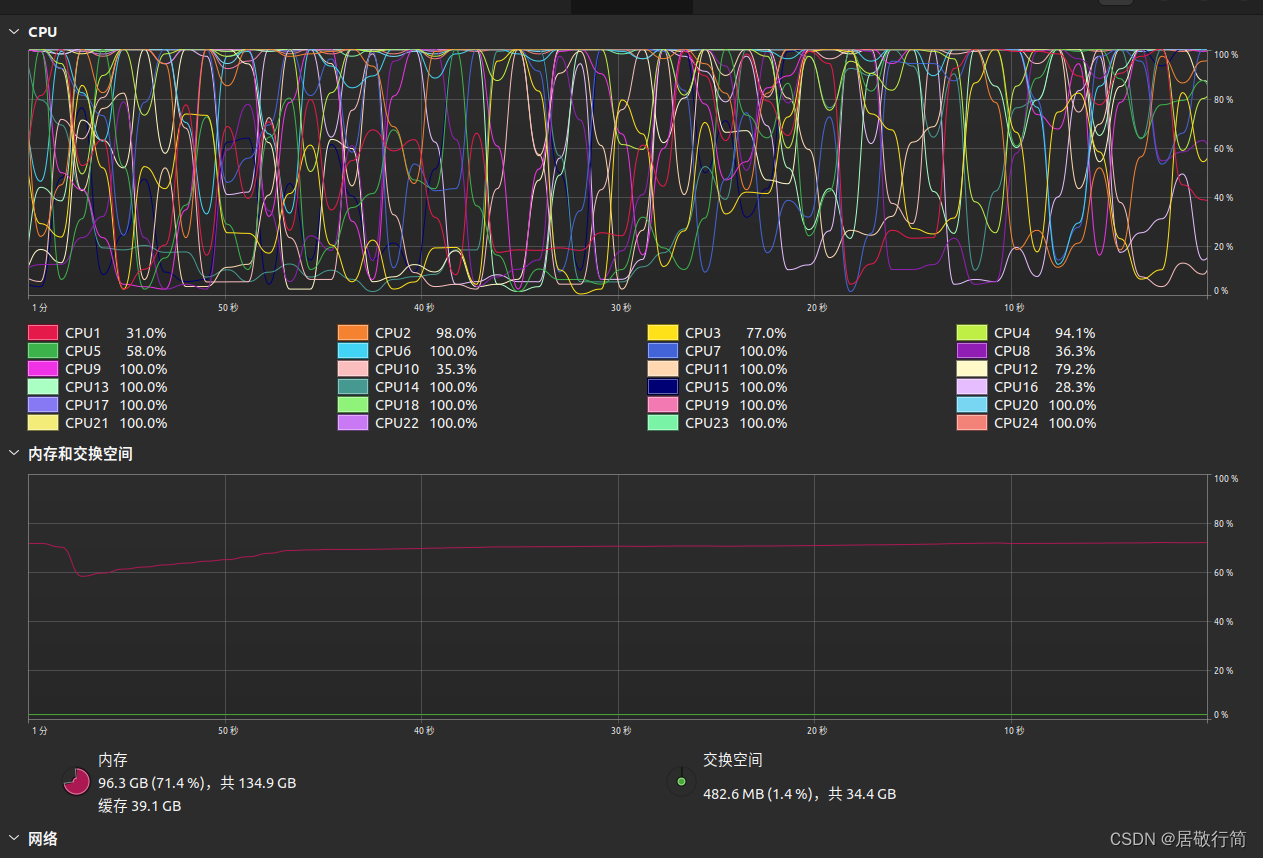
运行负载(设置的32G swap根本用不上)
结果分析
其他的文件都不重要,结果文件夹里out文件夹的内容是我们需要的
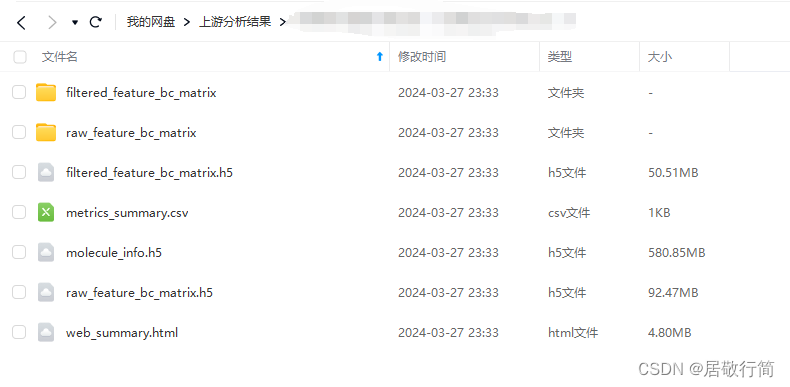
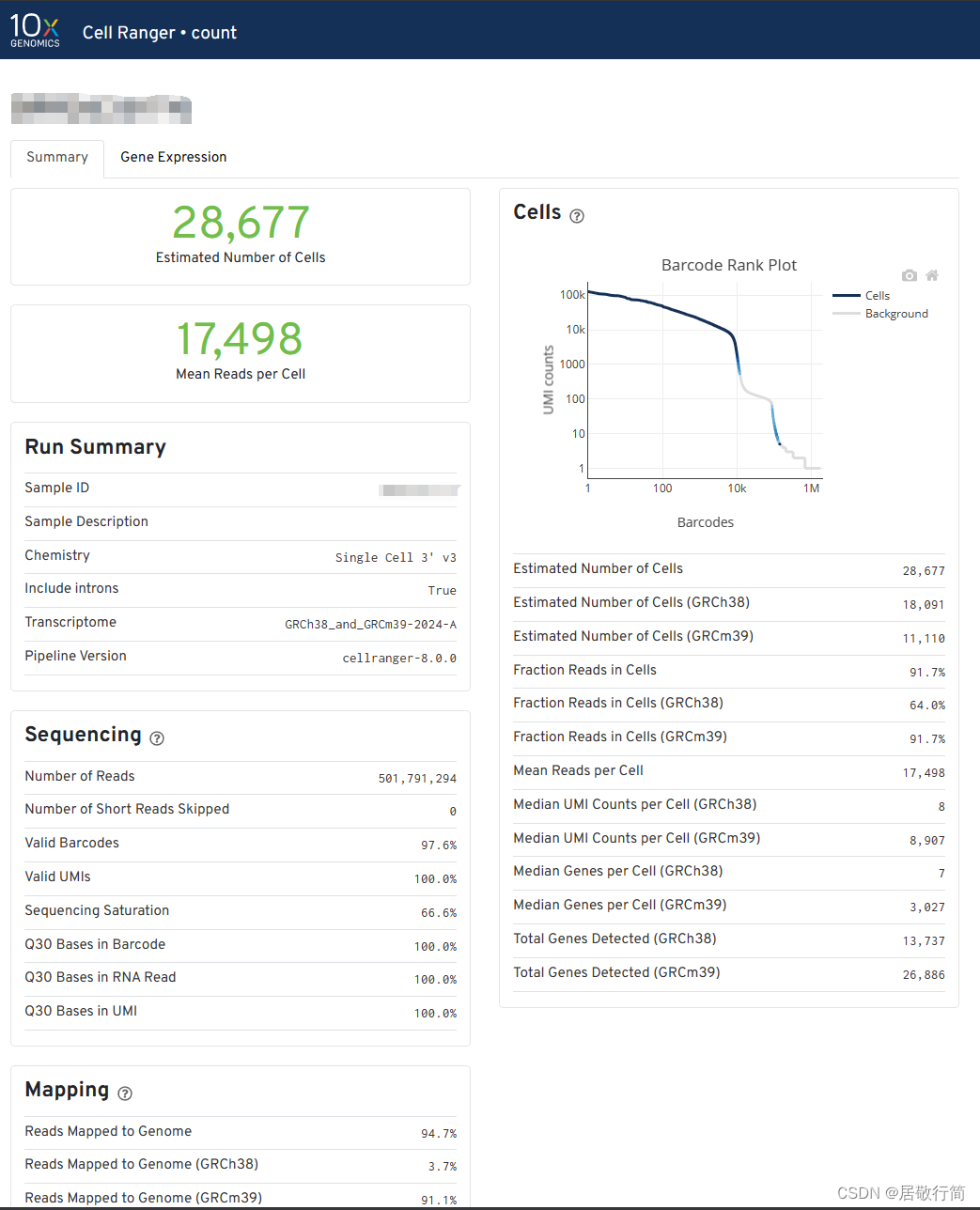
最下面那个html是对流程的概述,可以看一眼。
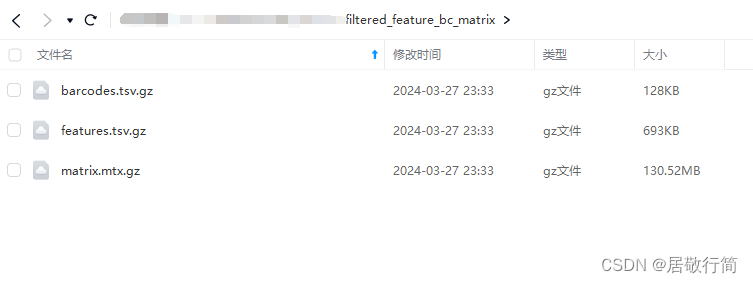
我们下游分析文件在filtered_feature_bc_matrix文件夹里
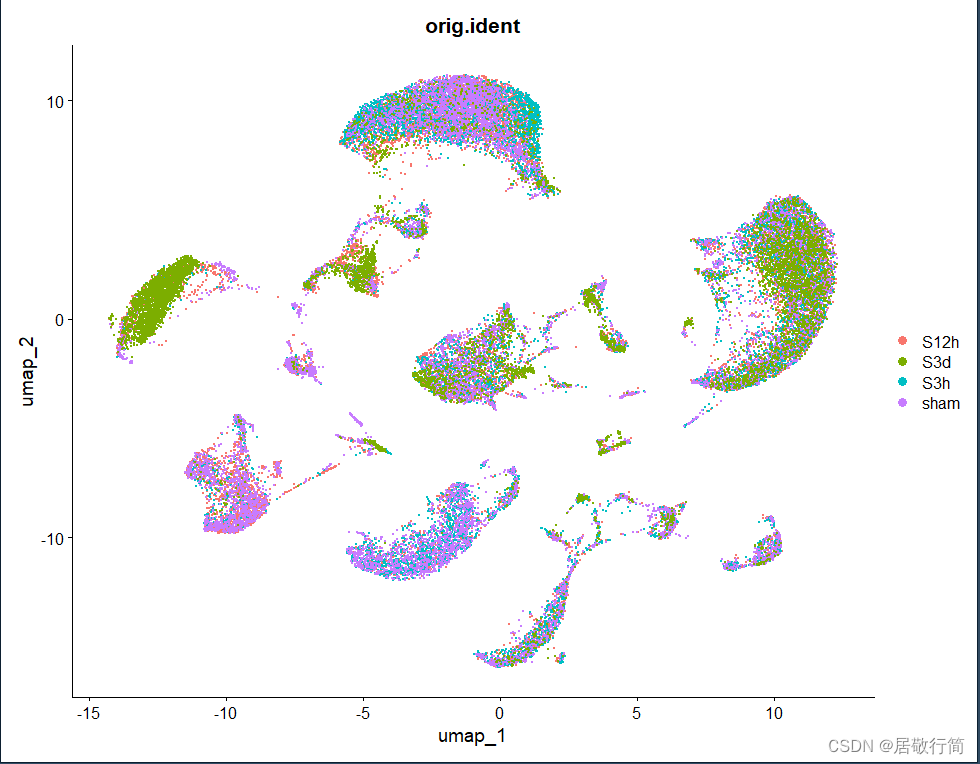
下游降维聚类结果:

















 5984
5984

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









