










1、体系的总能量---内能
一个模拟的体系,无论是NPT还是NVT,都有总能量,就是内能。
内能=动能+势能
势能=键能+非键作用能
对模拟过程的记录的能量变化进行绘图,可以检查能量是否达到收敛
2、结合能、熵能和结合自由能(亥姆霍兹自由能)
假设在真空中,有较远距离的分子A和分子B碰撞后结合在一起,形成新分子C,C的能量低于原先能量的加和。这时释放的能量就是结合能(前后内能的变化ΔU)。
熵值的大小和体系的混乱程度有关,体系越混乱,熵值越大。熵值不等于熵能。
熵值(ΔS),熵能(-TΔS)
体系是要自发向混乱程度增大的方向发展的(这是神明大人喜欢的),我们就理解为混乱程度的扩大是熵能的释放。
例如,将一些磷脂分子丢进水里,磷脂分子头部亲水,尾部疏水,会自发的聚成双层膜的球体,这个过程体系由无序变的稍微有序(可以想见原本有更多种运动可能的磷脂分子的运动性能减弱了),体系的熵值减小(ΔS),相反的体系的熵能(-TΔS)要增大。
但是分子动力学不能模拟熵
体系的自由能变化就是体系的内能变化加上熵能变化:ΔA=ΔU-TΔS
3、吉布斯自由能
结合自由能是适用于恒容(NVT)的体系,这样的体系体积不用,忽略了外界的压力做功。
对于恒压(NPT)的体系,这样的体系体积会变化,需要考虑外界的压力影响。
考虑体积变化的体系的自由能称为吉布斯自由能:ΔG=ΔU-TΔS+PΔV
4、真空中如何计算两个分子的结合自由能
利用吉布斯自由能格式:ΔG=ΔU-TΔS+PΔV
在真空中,压力P为0
设置温度T=0K,这时熵能-TΔS也为0
在0K,0Pa的环境下,结合自由能,就会等于结合能(ΔG=ΔU)
等温等压下,不做非膨胀功条件下的自发过程,一定是自由能减少的方向,这是热力学第二定律的一种表述的形式。
5、在分子模拟中计算ΔG的方法:
在分子模拟中,熵的变化不能捕捉,即不能通过公式ΔG=ΔU-TΔS来求ΔG
但是在实际实验中可以根据ΔG=-RTIn(K)进行计算
这里R和T都是常量(R表示气体常数 R=8.314J/(mol*k)),In是以自然数e为底的对数,K是反应的平衡常量。根据反应物和产物的平衡浓度得到,K=C(产物)/C(底物)
若是相比底物,产物的平衡浓度越大,则K值越大,底物的自由能越低,ΔG<0
模拟过程中如何得到这个K:
K,反映的是一个由自由能落差而产生的两种状态之间的分布状态不同,就是概率密度。
例如,一个轨迹中,构象有两种RMSD,在RMSD-1停留的时间占模拟时间的0.4,则RMSD-2的占用时间比例就是0.6,K=0.6/0.4
代码计算下这个ΔG:
import math
#计算平衡常量:
lnK=math.log(0.6/0.4,math.e)
#转换R的单位j/(mol*K)-->kcal/(mol*K),这里1kJ=0.2389kcal
R=8.314*0.2389*10**-3
T=310
#计算自由能:
G=-R*T*lnK
print(G)得到的结果是-0.24965562242965442kcal/mol
6、反应坐标、概率密度分布和平均力势
凡事变化的过程,例如丙氨酸三肽的顺反异构、一个小分子通过一个通道蛋白,这些过程都可以用一些特定的分子坐标从头到尾串联起来表示,这些特定的分子坐标就称为--反应坐标。模拟过程中,每一步算一个采样(分子坐标),这些坐标不是都能作为反应坐标。
在amber的A17教程里,对丙氨酸三肽的15号C和17号N之间的酰胺键进行180度的旋转。我们设置间隔度数为3度,制作出61个状态的反应坐标,对连续的线状反应坐标离散化(变成61个点)
对每个状态进行相同步数的模拟,计算各个状态中体系位置的分布概率,统计出的曲线就是概率密度分布。将其转化为自由能势能面(Potential of mean force,PMF),也叫平均力势
所谓的势能面,就是反映能量变化的曲线或曲面。自由能势能面就是反映自由能变化的曲线。
问题:关于概率密度分布如何转化为PMF
概率密度分布也叫做径向分布函数(Radial Distribution Function,RDF)
了解RDF的概念:
对一个参考的粒子,测量在以其为中心,半径为r的圆面内出现粒子的概率g(r)
归一化后的公式g(r):
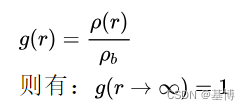
p(r)为距离A粒子 r 处B粒子的密度
pb为距离为所有以A离子为中心, 半径 rmax 的壳层内B离子的平均密度
PMF的概念:
为在给定距离下,将两个粒子从零相互作用状态可逆地带入完全相互作用状态所做的功:W(r)
从g(r)得到W(r):
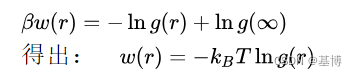
这个公式也叫做Kirkwood-Buff(KB)解理论
7、伞形抽样
问题:模拟时间有限,很难采到高能垒的构象
我们的模拟体系,在很多时候都老老实实的呆在能量低的构象里不思进取。我们希望模拟过程中体系可以出现的能量较高的构象,但在实际的模拟中出现的概率很小。
能量高的构象,模拟体系爬不上去,那真实的体系就能爬上去吗?。。。能,一个构象能量高,不是说永不可及,而是概率低,但只要时间足够长,体系就能爬上去呆一会儿。所以有些真实的反应需要1~2天,中间需要穿过很高的能垒和发生概率很低。
但对于模拟2~3周才有几个ns的模拟,这样跑出的体系很难访问到反应坐标上的高能垒部分,采样的范围受限。
解决这个问题的就是伞形抽样(Umbrella Sampling,US)
Umbrella Sampling,firstly proposed by G.M. Torrie, J.P. Valleau from Canada at 1997
如果我们的体系不愿意爬上高能量的反应坐标,我们就用力强迫这些原子到这些高能量的位置,具体的做法是添加一个弹簧势能w=k(x-x0)^2(偏置势,Wi),在反应物上,将反应物绑在高能状态。要是限制的原子跑出这个限制的范围,弹簧就会给出一个往回拉的力,将原子拉回范围内的位置。
例如在amber教程A17中,对丙氨酸三肽的15号C和17号N之间的酰胺键添加一个简谐势,强制这个酰胺键在高能的位置。
我们将一系列反应坐标分为一个一个小窗(window),就是变化的构象(酰胺键从0度~180度,间隔为3度的扭转),在每个构象都跑三连模拟(最小化、弛豫和成品)。
对第i个窗口进行采样,得到的RDF值称为有偏概率Pib,根据这个概率计算得到第i个窗口的无偏自由能Ai(去掉偏置势的自由能为无偏自由能)。
为什么叫伞形抽样
由于偏置势多是1/2K(Z-Zi^2)这种二次函数曲线,图像就像一把伞一样覆盖窗口,故 取名“伞状采样”。
经过后处理方法,合并窗口,得到整个反应过程的无偏自由能Au
8、加权柱状图分析法(Weighted Histogram Analysis Method, WHAM)
一个实现在伞形抽样中将有偏采样转化为无偏采样的方法
WHAM的大致过程:
1、计算每个窗口的无偏概率密度Pi(x)
无偏概率密度就是偏置势Wi=0的真实RDF
2、对各个窗口的概率密度Pi(x)进行线性组合,得到每个反应坐标的整体P(x)
3、根据P(x),和公式A(x)=-kT*lnP(x) 计算各个反应坐标点的自由能,绘制PMF曲线
9、相空间的概念
一个随时间演化的system, 我们可以描述出它的运动轨迹, 以及它在运动轨迹上任一点的速度, 这样它的演化过程就被完全描述. 所以, 我们可以用它在每个时刻的position和velocity来描述它在任一时刻的state. 在 classical mechanics当中, 我们认为一个system在某一时刻的position和momentum 也是是唯一确定的, 因此, 这个system随时间演化的过程中所处的每一个state 也可以由系统的position和momentum来描述. 假定有一个在三维空间中运动的粒子, 它在任意一个时刻都有三个coordinates x , y, z以及三个momentum components px, py, pz, 所以我们要描述这个单粒子在某一时刻的运动状态, 就需要6个分量. 按照惯例, 我们将位置坐标记为q, 动量坐标记为p, 这个pq空间就称作phase space. 显然这个相空间是一个六维的空间, 其中动量分量和坐标分量各占三维. 如果一个系统处在三维空间, 这个系统包含N个粒子, 那么我们为了在phase space中描述这个系统, 就需要一个6N维的phase space. Phase space 中的每一个点就表示系统的一个state.
内容参考:



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











