







数据回帖
根据维基百科的定义:在计算和数据管理中,数据映射(data mapping)是在两个不同的数据模型之间建立数据元素映射的过程。
一个经典的pattern mapping问题:查找pattern(P)中字符串(T)的重复次数。通常的解决方法是使用后缀树,在之前的文章中写过方法:后缀树练习实例:从目标串S中查找串T重复次数
在生物信息中,根据有无已知的基因组信息可以将mapping分成两类。这里只谈mapping基因组信息已知的情况,即将数量庞大的基因片段(reads)与已知的基因组(reference genome)做比对。
(关于两种类型的介绍可以参考Read mapping or alignment (EMBL-EBI)

(关于质控的部分可以参考 Quality control (EMBL-EBI))
最朴素的算法就是历遍整个基因组,找到序列(reads)出现的次数。然而在基因序列的处理中显然是不可行的,因为通常基因组的数据长度达到数十亿,reads的长度通常也有100bp,计算量相当庞大。
因此通常采取两种策略:
- reads的预处理
- 建立一个哈希表,其中包含reads中存在的所有K-mers
- 每遍历一次基因组,增加相应的kmer数量加一
散列表(Hash table,也叫哈希表),是根据键(Key)而直接访问在内存储存位置的数据结构。
在生物信息学中,k-mers是生物序列中包含的长度为 k k k 的子序列。
- 基因组数据的预处理
- 建立index:将一组基因组序列切分成若干seed片段,每一个小片段给标注一个index,通过index可以查找到该段基因而无需调用整个片段。常用的index方法是哈希算法:给每一个片段计算Hash值作为其在索引表中的地址。并在地址表中保存这段序列及其在基因组中的坐标,这样在reads中再出现这个片段的时候我们就能在地址表中迅速地找到其在基因组上的位置。
- 通过索引(index)查找seed hits的方法:Prefix Tree and Suffix Tree
好处:合并共享子串,降低内存消耗,方便最长子串的查找 例如BWA等利用Burrows-Wheeler transform (BWT),BWT是基于后缀的转换,可以逐位对比并延申片段 (BWT算法推导:20171026-基于BWT算法的比对软件原理解析(BWA & Bowtie & Bowtie2)
一般的回帖工具(如bwa,gem等)采用第二种策略。类比于文章开头提到过的数据映射经典问题,pattern P 就是基因组信息(reference genome),而字符串T对应reads,那么问题就变成了“查找基因组序列中某个基因序列(read)的重复次数”。
multi-mapped reads的处理
如果用正常思维来理解,某条序列要么没有map到基因组上,要么匹配到基因组某个位置上。但在现实中常常会出现一个序列map到多个染色体上的情况,出现的原因可能有:
- 基因组包含的重复序列(repeats)比读取的序列长
- 一个read显然一次只能来自一个位置,但如果如果重复序列的两个副本都被测序,那么就会有两个reads。
基因组包含的重复序列比读取的序列长的情况
- 使用更长的reads
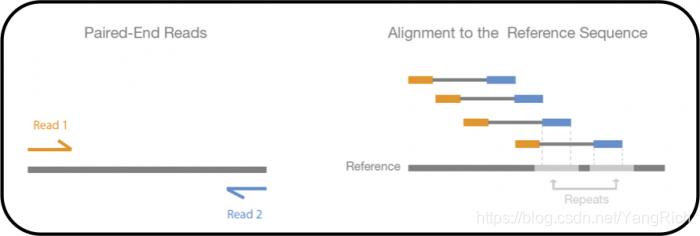
- paried-end reads :
配对末端测序可对DNA片段的两端进行测序。 因为每个配对读段之间的距离是已知的,所以比对算法可以使用此信息在重复区域上更精确地映射读段(Design considerations - Library preparation (EMBL-EBI))。成对的末端读取减少了多重映射的问题,因为一对读取必须在一定距离内且以一定顺序映射。
- 长读长测序技术(例如Pacific Biosciences的SMRT,Single Molecule, Real-Time (SMRT) Sequencing is the core technology powering our long-read sequencing platforms.)可以获得足够长的reads长度,可以对大多数基因进行完整的转录本测序。
其他情况
4种处理策略:
- 只采取第一条match的结果(Bowtie)。缺点:总是会选择chr1(因为是按照数字顺序排序…)
- 采用所有结果(GEM)。缺点: This will due an over-representation of the loci abundances, and actually is against the assumption of all packages that perform differential expression in count data.
- 选择任意一个结果(MAQ)。缺点:有可能刚好选择了错误的那个结果…
- 放弃所有结果。缺点:显然会丢失大量数据信息,低估基因家族内所有基因的表达水平
根据数据的来源(DNAseq还是RNAseq),可以采取不同的策略:
- DNAseq:将reads随机分配到某一个位置可能是合理的,但隐含着所有重复片段的覆盖范围相同的假设。
- RNAseq:选择任意结果的方法不太合理,因为在一个基因家族中,一个copy可能比另一个copy表达水平更高(即reads数量更多)。

















 882
882

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









