








关键词:多组学;基因测序;变异检测;
文献简介
- 标题(英文):Integrating WES and RNA-Seq Data For Short Variant Discovery
- 标题(中文):整合 WES 和 RNA-Seq 数据以进行短变异发现
- 发表期刊:(预印版暂无期刊)
- 作者单位:Sequentia 生物技术有限公司
- 发表年份:2021
- 文章地址:https://doi.org/10.21203/rs.3.rs-847277/v1

图1 文献介绍
组学的整合具有巨大的潜力,可用于变异发现。已经开发了几种算法来以整合方式检测体细胞变异,但仍然没有胚系突变检测的策略。在此基础上,研究者开发了一种通过整合WES和RNA-seq数据来识别胚系突变的策略。这种整合策略从原始序列数据中识别短变异(SNP和插入缺失),将其分为六组以改善变异解释:强证据,仅DNA,仅RNA,等位基因特异性表达(ASE),RNA编辑和RNA挽救变异。研究者基于整合流程分析了四个样本,与仅使用 WES 数据相比,发现已识别变体的数量有所增加,但对性能没有太大影响,从而可以验证由两种类型的数据(强证据变体)识别的变体,并鉴定 RNA 编辑和 ASE。这种整合策略提供了一种从WES和RNA-seq数据中鉴定胚系SNP和插入缺失的方法,充分利用这两种组学可以扩大已识别变异的范围并进行变异验证。
在该整合分析流程中,研究者将Sentieon 工具中各模块进行了深度整合利用。Sentieon 工具的胚系突变发现流程为 DNA 和 RNA 层面的突变精确、快速检测提供了支持。
测序流程
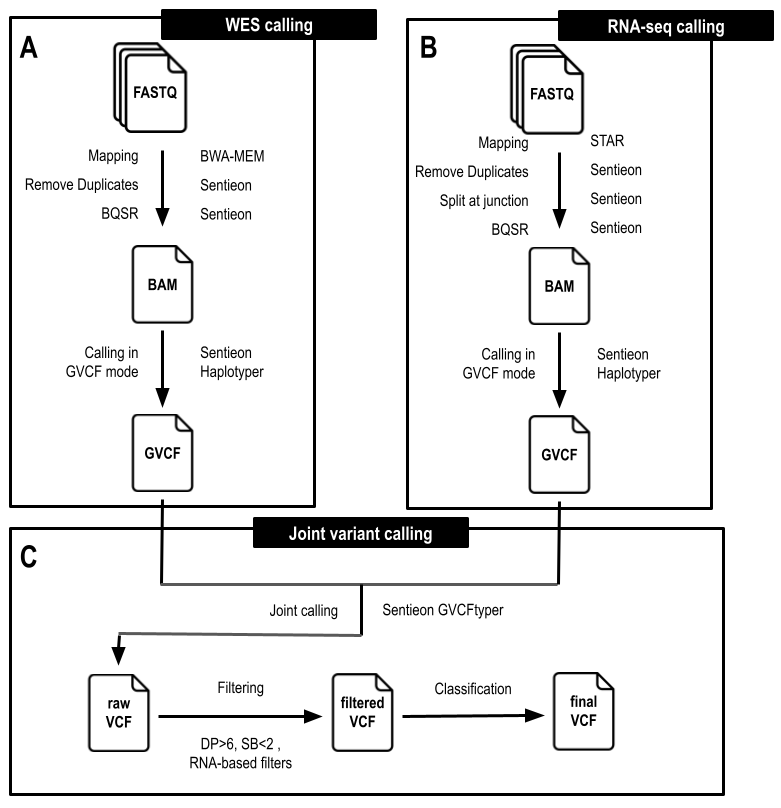
图2 使用 WES 和 RNA-seq 数据进行变异鉴定的工作流程。工作流程主要包括三个步骤:A) WES 比对和变异检测。B)RNA-seq比对和变异检测。C)WES和RNA-seq数据的联合基因分型,变异过滤和分类

图3 Sentieon 的作用
Sentieon软件团队拥有丰富的软件开发及算法优化工程经验,致力于解决生物数据分析中的速度与准确度瓶颈,为来自于分子诊断、药物研发、临床医疗、人群队列、动植物等多个领域的合作伙伴提供高效精准的软件解决方案,共同推动基因技术的发展。 截至2023年3月份,Sentieon已经在全球范围内为1300+用户提供服务,被世界一级影响因子刊物如NEJM、Cell、Nature等广泛引用,引用次数超过700篇。此外,Sentieon连续数年摘得了Precision FDA、Dream Challenges等多个权威评比的桂冠,在业内获得广泛认可。
文献讨论

图4 文献讨论
RADIA工具侧重于利用RNA-seq数据来验证DNA测序中发现的体细胞变异,确保所有报告的变异都有DNA层面的证据支持,排除了仅基于RNA数据的变异调用,从而忽略了RNA编辑等特定变异类型。而VaDiR则采取相反策略,主要依赖RNA-seq数据来识别体细胞变异,DNA数据仅用于过滤掉种系变异,这种做法可能导致低频RNA-seq变异的遗漏,且未能充分开发结合两种数据类型以发现更多生物学意义的变异。
总结
综上所述,研究者提出的胚系突变整合方法增加了检测到的变体的数量,而对性能没有太大影响。整合方法还允许验证由两种类型的数据识别的变异,并通过将它们分为六组来改善变异的优先级:强证据、仅 DNA、仅 RNA、ASE、RNA 编辑和 RNA 挽救变异。















 263
263











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









