






诊断和治疗异质性脑部疾病需要具有跨遗传学、蛋白质组学和神经成像多个学科的专业技术。NeuroPM-box旨在满足这一需求,它是一款用户友好、开放访问、多工具的跨平台软件,能够描述多尺度和多因素的神经病理学机制。该工具包使用先进的分析模型进行分子、组织病理学、脑成像和临床评估等多种应用。通过体外(N > 2900)、体内(N = 911)和尸检(N = 736)神经退行性样本数据(包括临床特征)验证了:
(i)涵盖数十年疾病进展的一系列连续状态(遗传、组织病理学、影像学或临床改变);
(ii)病理因素(如淀粉样蛋白、tau 和α-核糖蛋白)同时在脑内传播;
(iii)多种生物因子之间的协同相互作用(例如,毒素对脑萎缩的影响);
(iv)基于疾病异质性和治疗需求的患者分类。这个k工具箱(http://neuropm-lab.com/neuropm-box.html)可以有助于了解复杂的大脑过程,并加速神经病学中精确医学的实施。本文发表在COMMUNICATIONS BIOLOGY杂志。
背景介绍
最常见的神经系统疾病是高度复杂的,涉及从分子到宏观(系统)水平的一系列生物学变化。例如,阿尔茨海默氏症(AD)是最常见的痴呆症形式,其特征是基因、分子通路、蛋白质、血管、神经元突触和高阶神经网络同时中断。这些因素和其他因素之间的共同作用,而不是单一的主导因素,是导致AD在记忆、思维和行为方面相关变化的原因。AD单靶治疗干预的失败清楚地表明,如果不对其众多相互关联的因素进行更深入的研究,我们无法理解或最终治愈复杂的多层次脑部疾病。根据个性化医学(PM),需要针对多尺度和多因素的大脑机制以及每个人的反应能力进行定制治疗。
在过去的几十年里,科学界越来越了解对大脑的相关疾病进行综合(多层次)分析的必要性。系统生物学旨在生成分层生物网络的时空机制模型,以及大脑从正常状态向病理状态转移时的适应性变化。神经信息学领域同样致力于开发分析和计算模型,用于共享、整合和分析多模态神经科学数据。然而,尽管它可能更好地了解复杂的神经病理过程和个性化的治疗选择,但大多数相关方法(例如分子-神经成像分析、数据驱动的患者分级和脑内病理改变)仍然难以应用,即使有共享计算代码,通常也需要先进的编程技术,在许多情况下,甚至需要开发人员的合作。简言之,对于多尺度和多因素大脑研究而言,仍然缺乏重要的用户友好型开放访问工具。由于需要这些类型的工具的创新方法加速发展,这助长了统计上的不一致,消耗了宝贵的研究经费,并且仍然是研究重复性的主要障碍。
在这些担忧的激励下,我们开始了一项长期举措,开发、验证和共享分子、组织病理学、脑成像、认知行为和治疗数据的综合分析模型,以增进对个体和群体层面大脑(分解)组织机制的理解,以及识别个性化的治疗需求。随后,我们开发了用户友好型软件,大大改进和统一了多种方法在一个单一的应用程序:神经信息学个性化医疗工具箱(NeuroPM-box,见图1和表1),可以不受限制地应用于任何类型的神经科学数据。例如,这些工具中的每一项都经过了广泛的测试和验证,但它们同样适用于在健康的神经发育和衰老或心理障碍中描述多因素过程。工具中的大部分输出在生物学上是可解释的,4Dviewer能够可视化大脑的多因素时空动态(例如,tau 和淀粉样β蛋白通过皮层传播)。此外,NeuroPM-box不是静态应用程序:它旨在通过新的、更综合的方法不断扩展,以加速对异常大脑机制的理解,并推动神经病学个性化护理的实施。

图 1 NeuroPM-box软件工作流程和实用指南示意图
a 输入软件的数据包括分子(如RNA和蛋白质浓度矩阵)、多模态成像(如tau、淀粉样β和葡萄糖代谢PET、血管、功能和结构MRI)、全脑连接组学(如结构和血管网络)、认知/临床评估和治疗干预(如药物治疗)。对可以分析的数量没有限制。
b NeuroPM-box允许用户从四种分析方法(工具),应用辅助程序,访问可视化工具(NeuroPM-viewer)和软件教程中进行选择。
c 支持多模态数据驱动分析的主要软件模块。
d NeuroPM-viewer能够详细探索人类皮层的真实和建模空间的大脑动力学。
e 用户指南。从本质上讲,分析方法分为两大类:经验方法和机械方法。前者纯粹是数据驱动的,侧重于识别和解释数据中的内在模式,而不做生物学假设。具体而言,所包含的算法提供反映疾病进展的个性化定量分数,并将每个受试者分配给不同的亚群体。任何类型的定量数据都可以用作输入(如转录、蛋白质组学、组织病理学、代谢学、多模态成像、临床),而每个数据特征对受试者最终分层的贡献都是量化的,揭示了最详实的特征(如特定基因、大脑区域、临床评估)和相关数据模式(如RNA、成像、临床)。但是,由于区分直接和间接生物效应的内在局限性,用户应避免基于实证建模进行因果解释。相反,机械模型旨在根据生物因素的变化来解码因果关系,这些变化通过大脑连接和/或协同因子——因子相互作用而传播,从而促进时空大脑重组。两个已实施的生成模型侧重于单模态或多模式成像数据,即 ESM(流行病传播模型)考虑用特定成像方式测量的独特生物因子的脑内分布(例如 tau-PET,或淀粉样蛋白-β PET),而MCM则考虑多种生物因子的直接相互作用和并发的脑内扩散,这些变化与不同的成像方式(tau、淀粉样蛋白-β和葡萄糖代谢PET、脑血管流动、功能活动指标和用MRI测量的结构萎缩)相量化。值得注意的是,机械方法(ESM、MCM、pTIF)可以通过经验数据驱动输出(cTI分层)获得信息,从而允许在基于成像的生成大脑模型上纳入各种可能的多尺度生物信息(如分子和临床阶段和亚型)。最后,通过多模态机械方法确定个性化因果脑模型,以确定个体治疗需求,即停止/恢复因子特异性(成像模式)或临床恶化。
表1主要NeuroPM-box的方法及其协同作用
结果
NeuroPM-box(图1)能够对大规模分子和宏观数据进行单独和组合分析,包括分子筛选(转录组学、蛋白质组学、表观基因组学)、组织病理学(死后神经病理学)、分子成像(淀粉样发射断层扫描PET)、磁共振成像(MRI)和认知/临床评估。特别是,它侧重于澄清关于大脑如何运作的关键机械问题:如(一)哪些系列的连续分子或宏观状态(如遗传和大脑区域变化)是数十年神经病理进化的基础?(二)哪些基因(或分子通路)驱动其他基因和通路功能障碍?(三)疾病因子(如毒性蛋白和淀粉样-β蛋白质)如何通过大脑中的细胞传播?(四)病变脑区发生了哪些多因素、协同(因果)相互作用?(五)每个患者对不同的治疗干预措施有何潜在反应?
在多个研究中开发并成功验证了所包含的每种工具和分析方法。NeuroPM-box聚合了这些工具的显著改进版本。值得注意的是,它首次允许不同方法和数据模式的协同组合,实现统一的多尺度和多因素大脑分析。图1e和表1总结了不同的算法,并提供它们进行联合分析的示例。简言之,用户可以访问四个主要分析框架、其中的组合和多功能可视化工具。大规模分子、成像和/或临床数据的轨迹分析。
追踪大尺度分子、成像和临床数据的轨迹
对比轨迹推理(cTI)算法(图2 cTI定义中的"在线方法")使用人工智能(AI)的最新进展来探索和可视化高维数据,使用大规模生物观测(例如,涵盖相关生物过程的遗传学和神经成像数据,如神经发育或神经退行性变),阐明独特的基础通路。例如,应用cTI 算法从 744 名阿尔茨海默病谱受试者的血液中发布基因表达(GE)数据,自动识别涵盖数十年疾病进展的一系列连续的分子状态(例如基因改变),随后检测与这些模式一致的个体相对顺序。每个受试者获得分子疾病的评分,反映个人在已确定的长期疾病发展“时间线”上的位置。同样,当在1225例死后大脑中评估AD和亨廷顿病(HD)谱(HBTRC)评估时,cTI强烈预测了神经病理严重性和合并症(Braak,淀粉样蛋白和Vonsattel分期;HBTRC的结果见图2d-e)。
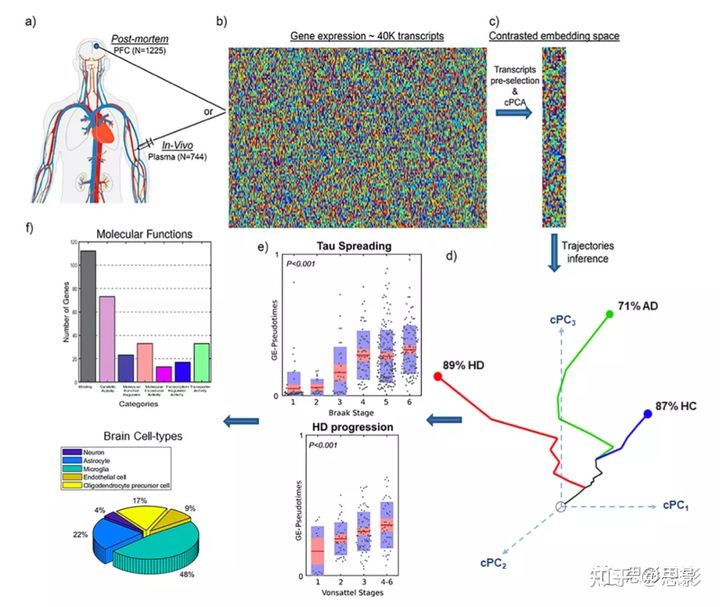
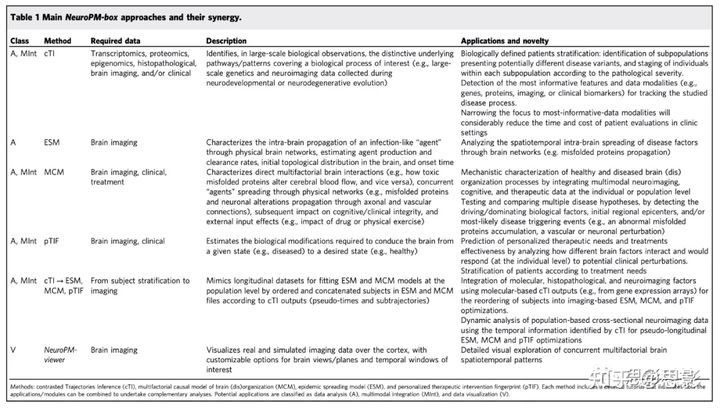
图2cTI(轨迹推理)应用示意图,以检测神经退行中与疾病相关的模式和患者神经病理学阶段。
a来自阿尔茨海默症(AD)、亨廷顿舞蹈症(HD)和/或正常对照(HC)受试者的体内血液(N = 744;ADNI)和验尸后的大脑(N=1225,ROSMAP、HBTRC)组织筛查,以测量约40000份转录组水平。
b通过对比主成分分析(cPCA),将每个人的高维数据简化为一组与疾病相关的成分。
d这允许每个受试者在减少的n维疾病相关空间中表示,其中相应的位置反映其病理状态(接近左下角意味着无病理状态;相反,右上角意味着高级病理学)。例如,在分析来自 HBTRC 高度异质人群(包括 HC、LOAD 和 HD,总N = 736)的GE数据时,高维数据降维到七个cPC捕获高达 97.5% 的人口学差异(和单独解释38.73%差异分别为19.91%、16.18%、8.46%、5.85%、5.50%和2.87%。请注意,为了可视化简单性,此处仅表示前三个cPC,但定量分析考虑了所有已识别的cPC。在此 cPC 内,每个受试者会自动分配到疾病轨迹,该轨迹表示可能遵循常见病变种的受试者的亚组(参见"方法")。亚组数量(疾病轨迹)根据受试者在疾病相关空间中如何"聚集"在一起自动确定。
e然后计算单个分子疾病评分,反映每个受试者在疾病轨迹中的进展程度。这个分数显著预测神经病理恶化。最后,生成的模型权重(来自对比 PCA)允许对最具影响力的基因/特征进行识别和后功能分析。
在HBTRC数据中,我们观察到(图2e)单个分子疾病评分与AD和HD神经病理影响水平之间的正相关关系,这与ADNI和ROSMAP的发现一致。GE(血液中发布基因表达)得分与Braak阶段和Vonsattel阶段显著相关。为了表征疾病异质性,cTI还可以将受试者分配到对比空间中的不同子领域。这些子项目反映了对比数据中的不同趋势,例如不同的疾病变异。为了验证 cTI 算法区分高度异质人群神经系统疾病的能力,分别重新分析了来自 HBTRC 的 GE 和组织病理学数据,包括两种疾病(晚发性 AD [LOAD] 和 HD)和非痴呆对照(数据集 1, N = 736;“Online methods”)。使用每种数据模式(GE 或组织病理学),cTI 方法自动识别反映特定诊断亚群的多个子轨迹(图 2d)。例如,基于GE,子轨迹1包括87%的非校正对照组、18%的AD受试者和2.7%的HD受试者;子轨迹2包括71%的AD受试者、38%的对照组和12%的HD受试者;第三亚组包括89%的HD受试者、32%的对照组和25%的AD受试者。
同样,根据有限的组织病理学数据(只有25个广泛的指标),子轨道1包括91%的对照组、28%的AD受试者和4.9%的HD受试者;子轨迹2包括62%的HD受试者、57%的AD受试者和40%的对照组;第三亚组包括48%的HD受试者、26%的AD受试者和2.3%的对照组。此外,我们使用合成数据(N > 2900)在不同的人口特征下,特别是不同人口异质性和样本大小下,广泛测试 cTI 的表现。模拟研究确认了模型在存在噪音数据的情况下能够准确识别单个疾病阶段和亚型以及恢复生物标志物对预测的贡献。
总之,合成和真实数据的结果表明,cTI在疾病分期和变异方面是一种很有前途的患者分层技术,即使考虑了共病的神经系统状况和嘈杂的观察结果。此外,cTI 可能还可用于从大规模横截面数据中提取内在动态信息。如表所述1(应用cTI → ESM、MCM、pTIF),此功能在分析横截面神经成像研究时特别有用,就好像它们是纵向研究一样。即 cTI(或任何用户提供的患者分层)的单独伪时间和子项目可用于模拟纵向数据集,使ESM、MCM 和 pTIF 模型适应亚人群水平。
病原体的流行病传播
神经学背景下的流行病传播模型(ESM)通过物理脑网络(如解剖、血管、功能)描述了感染因子(例如,错误折叠蛋白[tau、淀粉样蛋白、α-核素、TDP-43])在脑内的传播。ESM(图3a,b)估计了病原体的清除率和产生率,这与模拟感染的局部浓度呈线性关系。 ESM已被成功应用于进一步了解有毒淀粉样蛋白和tau蛋白在神经退行性人类大脑中的传播。重要的是,与初始模型应用程序相比,ESM 模型的NeuroPM-box工具包具有五大显著改进:
(i)数学扩展用于处理直接成像信号(例如PET的SUVr值)或图像中的概率推断值(原始模型仅针对概率值定义)。
(ii)通过考虑所有可用的时间点,改进后的ESM可以优化基于个体和群体的纵向数据(原始模型仅针对横向数据进行评估),从而提高模型估计参数的稳健性和生物解释性。
(iii)ESM使用更稳健的算法(MATLAB的MultiStart)来解决非线性微分方程。具体而言,基于梯度的算法从多个起点查找局部最值,以寻找全局解决方案。这种修改有效地提高了估计生物参数的重复性和解释性。
(iv)流行病的产生率和清除率可定义为线性或指数/S形函数(可选),通过考虑区域水平的线性和非线性生物过程提高了基本模型的灵活性(初始模型只考虑指数化产生率和清除率)。
(v)考虑到不同生理因素和/或随机噪声对分析成像模式的影响,以及非零区域值不一定意味着所研究的因子(如淀粉样蛋白或tau沉积)的存在,而只是图像信号的背景波动。随后,为了估计区域中心,改进后的 ESM (流行病传播模型)算法允许定义最大(非零)值,低于阈值,这些区域仍然被认为没有致病因子存在,但具有典型的背景噪音(例如 tau 和淀粉样体积阈值)。只有超过阈值的区域才被视为可能的扩散中心。在tau-和淀粉样-PET传播分析中,模型拟合性都有显著改善。
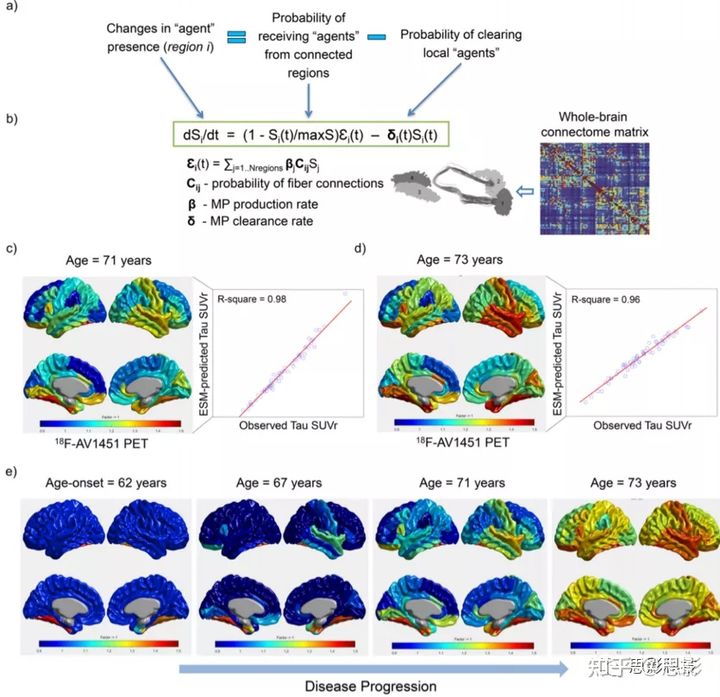
图3 ESM方法和脑内 tau 扩散的预测
a特定脑区中存在特定感染因子(例如淀粉样蛋白、tau 错误折叠蛋白[MP])。
b这种动态因果效应模型可在数学上转换为非线性微分方程组,该模型取决于单个因子的生成速率、清除率和大脑脑区连接矩阵。在NeuroPM-box中,可通过弥散加权 MRI 或替代技术估计脑连接矩阵。
c显示了在临床健康女性对照组的第一次18F-AV1451 PET评估(年龄=71岁)时,ESM结果再现了具有明显记忆问题的tau沉积模式(ADNI数据,受试者ID 024_S_5290)。
d、ESM 在第四次评估中(年龄=73岁)对同一参与者的结果。从无病理阶段开始,模型解释了四个可用时间点之间tau值86%(P < 10−10)的差异。
e、ESM模拟全脑脑内 tau 传播过程,从 tau 传播的估计发病时间到最后观察到的时间点。
在图3中,我们展示了改进的ESM算法对105名健康和患病人群的应用,每个被试至少有两次纵向tau-PET采集(18F-AV1451 配体、ADNI 数据)。平均而言,当在个体水平上应用时,此方法解释了80%(SD = 9.6,所有P < 10−6)的区域tau值的方差,包括所有可用时间点和受试者。图3c-e显示 EMS 的结果为:健康的女性对照参与者与重大记忆疾病和四个纵向 tau-PET 评估(ID 024_S_5290在 ADNI)。从无病理阶段开始,模型解释了四个可用时间点tau值变化的86%(P < 10−10)(图3c, d)。复制的tau沉积模式在第一次和最后一次PET评估时,71岁(图3c,右)和73岁(图3d,右)之间分别观察到显著差异。此外,ESM自动将左半球的内嗅皮质、梭状回、前扣带和顶下小叶确定为最有可能的传播中心,并估计tau积累和传播始于62岁左右。图3e说明了大脑内的长期传播过程,从已确定的传播中心开始,经过十年时间,按照与 AD 相关的模式扩散到其他大脑区域。在本分析中,我们允许最多四个区域作为初始中心,并允许在开始时间非中心区域的启动值最多为最大观测 SUVr的5%。
大脑组织的多因素因果模型
大脑组织和认知的多因素因果模型(MCM)(图4a,b)解释了:(i)疾病过程中的第一个病理扰动(最初改变的大脑区域和生物因子),(ii)多因素因果相互作用(例如,毒性错误折叠蛋白如何改变大脑血流[CBF],CBF的后续变化如何影响神经元活动和灰质萎缩,反之亦然),(iii)通过物理网络同时传播扰动(例如,错误折叠蛋白质的脑内传播,血管或神经元在血管连接上的改变),(iv)i和ii对认知/临床完整性的后续影响,(v)外部因素的特异性影响(例如,给定的临床治疗如何影响 tau、淀粉样蛋白和 CBF)。MCM(多因素因果模型)认为,一旦特定因素事件发生在给定的大脑区域或一组区域,它就可以直接与其他生物因素相互作用,从而改变其状态。这些改变也可以通过物理连接(如解剖、血管连接)传播到其他大脑区域,在这些区域中,类似的因子和传播机制可能在一个连续的周期内发生。MCM(多因素因果模型)已成功应用于 AD 研究,阐明了多因素疾病特异性机制,目前正在应用于其他神经退行性疾病(如肌萎缩性侧索硬化症、前额性痴呆症、帕金森病)。
图4 MCM定义和预测在临床前AD中并发的 tau、淀粉样蛋白和脑结构变化
a特定脑区给定生物因子(如淀粉样蛋白、tau沉积)的变化被建模为局部多因素协同作用的函数(如脑血管流动失调如何影响淀粉样蛋白和tau沉积),脑内变化通过通讯细胞和外部输入(例如治疗)传播。
b此动态因果效应模型可在数学上转换为微分方程组。类似于先前提出的脑功能因果模型,在MCM中,因果关系是其微分方程的固有特征。除了传统的单因素建模方法(通常神经元活性或折叠错误的蛋白质),MCM方程还描述了:
(i)给定的生物因子在给定的大脑区域中的当前状态如何导致自身或相同或不同大脑区域中的其他生物因子发生新的变化,通过多因素局部交互作用或通过大脑连接传播,以及(ii)大脑的动态物理系统如何因外部输入的影响而改变(例如认知/感官刺激、治疗干预、环境影响)。c MCM模拟了临床患有显著记忆疾病的健康女性(从 67 到 72 岁)被试脑内的 tau、淀粉样蛋白和灰质(GM)密度的同时变化。然后,为了预测目的,计算了一个额外的多因素数据的时间窗口(从73到76岁)。请注意,在大脑灰质密度大幅降低的同时,tau 沉积显著增加。在a中,FUNC 是指休息时的功能活动(例如,来自 fMRI),METB 是指葡萄糖代谢(例如,来自 FDG-PET)。
在这里,改进的 MCM (多因素因果模型)算法应用于 504 名健康和患病被试(ADNI 数据),每个被试都有4–6种不同的成像模式和至少四次纵向评估。成像方式包括tau-PET、淀粉样-PET、FDG-PET(用于量化葡萄糖代谢)、静息态-fMRI(用于静息态神经元活动)、ALS-MRI(用于脑血流)和结构MRI(用于灰质密度)。所有被试平均纵向时间窗为 5.1 年(SD = 3.6)。MCM 模型优化成功地融合了 98.4% 的被试(504中的496名被试)。从个人层面的无病理状态开始,MCM 分别解释了 92% (SD = 4.7,P<10−6)和71%(SD=15.1,P<10−6)所有个体的多模态观测结果。图4c以MCM为基础的新分析,对具有显著记忆力的临床健康女性对照的tau、淀粉样蛋白和灰质萎缩的脑内变化进行了分析。但这里的特点是从多因素的角度对其进行了描述(这是第一次从单变量tau分析到综合多模态表征)。在本研究中,对于第一个(67年)和最后一个观测到的时间点(73年),该模型分别解释了6种数据模式之间94.6%和51.9%的差异。数据模拟(图4c)在最后一个时间点之后再延长三年(从73年延长到76年)来说明 MCM 算法预测未来疾病进展的能力。在此特定情况下(图4c),我们看到在大脑灰质密度大幅降低的同时,tau沉积显著增加。
基于成像的治疗干预
个性化治疗干预图谱(pTIF;图5)假设异质人群中的患者需要不同的治疗,这取决于他们大脑中的单因素变化(例如,tau/淀粉样沉积与否,脑血管改变与否,萎缩与否)及其多因素大脑动力学:不同的生物因子如何相互作用,并有可能(在个体层面)对临床扰动做出反应。基于对多模式成像数据(PET、MRI、SPECT)的时空分析,个性化治疗pTIF 值是一组多变量指标,反映了停止特定大脑重组过程或将病情恢复到正常状态所需的生物变化。换句话说,pTIF可以将大量数据(例如数千个多模式脑成像测量)整合到简化的患者特征档案中,表示控制该被试重组过程(如疾病演变)所需的生物因子的定量修饰。使用老化和晚发 AD 数据(ADNI)的结果演示了 pTIF 算法如何将患者分类为与mRNA 对应的基于治疗的不同亚型。在预测个体GE改变时,多模式成像衍生的pTIF大大优于认知和临床评估。此外,pTIF(个性化治疗干预图谱)鉴定的患者亚组在血液中呈现明显改变的分子通路,支持识别不同的病理亚型,因此在研究人群中提供不同的治疗需求(图5c)。
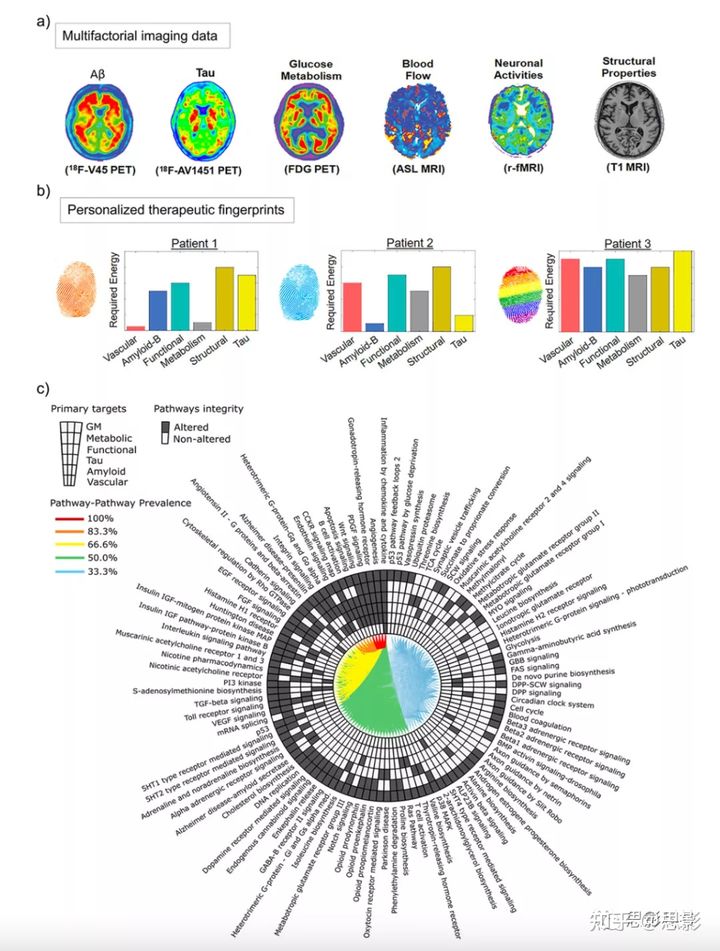
图 5 从多模式成像到治疗指纹和改变的分子通路(ADNI 数据)
a淀粉样蛋白、tau、CBF、静息态功能像、葡萄糖代谢和灰质密度的成像。基于网络的方法,pTIF 通过血管/解剖连接,使大脑内直接因子与因子的生物相互作用和多因素扩散机制进行个体表征。因此,pTIF 被定义为每个患者所需的一组变化。
b三个诊断相同的参与者的不同 pTIF 模式。患者1需要低成本的血管和代谢干预,患者2需要低成本的抗-Aβ和抗tau干预,这表明不同的单靶疗法可以分别使两个患者受益。然而,患者3需要多个单靶点干预,这表明高成本的组合治疗对这个病人更有利。
c改变的分子途径(血液数据)是不同单靶点治疗需求的基础。
可视化观察和模拟大脑时空动力学
更深入地了解正在研究的大脑过程需要获取和模拟大脑数据的可视化。NeuroPM-box包括一个多功能、用户友好的界面,可视化所有分析的大脑因素及其动态变化(图 S2)。时间是建模和可视化最重要的变量之一,因此,NeuroPM-viewer允许根据特定的可视化需求(许多其他设置)调整时间变量。
版本控制和稳定性
遵循共享计算神经科学软件的标准做法,我们采用先进的源代码管理工具进行版本控制(GitHub,https://github.com/)。此外,对每个软件版本进行的修改都会在软件网页上的.doc文件中注释并提供给用户,包括直观的技术解释,还讨论了相关的生物学影响。此外,不仅要用先进的信息学技术来计算,而且要提供稳定的结果。已在多个具有不同功能和操作系统(Linux、macOS、Windows)的工作站上成功确认了所实现方法(cTI、ESM、MCM、pTIF)的稳定性。
NeuroPM-Box教程
《软件用户指南》提供了所有可用的功能和选项的深入解释。它包括安装(Linux、macOS和Windows系统)的分步说明、数据组织标准化、特定于模型的输入和优化、离群值校正、数据可视化以及输出描述和解释。教程可从NeuroPM-box("Theory and Help"图标)和 https://www.neuropm-lab.com/neuropm-box.html 获得PDF版本。此外,还提供了用于 cTI 测试/评估(图 S1)的合成数据和实际演示脚本。
讨论
NeuroPM-box是一款跨平台、开放访问、用户友好的软件,用于使用目前存在的高级数学模型集成大规模分子、宏观和临床数据。NeuroPM-box允许分离和组合分析来自分子筛选(转录组学、蛋白质组学、表观基因组学)、组织病理学(神经病理学)、分子成像(淀粉样蛋白、tau-PET)、宏观 MRI 成像和认知/临床评估的数据。大多数可用的工具只关注分子或脑成像分析,而不是他们的综合分析,NeuroPM-box是专门为解决此类问题。此外,没有其他用户友好型软件包括表征改变效应(例如,以ESM和MCM为特征的连接介导tau和淀粉样蛋白的传播)的脑内扩散模型,或基于动力系统分析和控制理论(例如pTIF)识别个体治疗需求的模型。尽管一些在神经病学背景下基于生物标记物的患者分层的验证计算代码已被共享,但用户需要编程技术才能应用它们。NeuroPM-box的用户友好型实现,结合最近对所包含方法的改进,将加速与其他团队发布的几种最近开发的方法的比较和潜在整合。重要的是,使用NeuroPM-box不需要先进的数学和/或计算知识,因为每个模型都用直观的生物学术语来描述。然而,它被故意设计成一个后处理分析软件,而不是一个预处理包。许多优秀的免费软件已包含在其中(例如,GEPAS,Bioconductor,SPM,FSL,ANTs,CIVET,FreeSurfer,MRtrix3,DSI Studio,BrainSuite),因此,基本分子和成像预处理(成像配准、大脑分割、质量控制)应事先完成。NeuroPM-box用户应具备将数字数据写入/读取到文本文件的基本专业知识,并且在使用大规模群时,能够将数据组织到所需的格式中。
NeuroPM-box是一个长期的,持续的协议。所包含的工具都在不断发展,特别是在改进其数字优化(研究领域的开放式领域)和结果的解释/可视化方面。新的工具和方法也在开发中,目标是进一步整合多尺度和多模态神经科学研究。未来方法的增加将侧重于继续衔接分子、宏观因素(如神经成像衍生生物标志物)和临床数据。例如,我们正在整合一种新颖的 MCM 方法,它提出了一个通用公式,整合了数百个里程碑基因的全脑转录数据与多种神经成像衍生的生物因子(即淀粉样蛋白,代谢和tau-PET,血管,功能和结构MRI)和个体临床信息。这种统一方法成功地验证了健康的衰老和AD人群,同时解释了数百个基因对区域宏观多因素效应的直接(因果)影响,以及随后的畸变(tau,淀粉样蛋白)在轴突和血管网络上的病理传播,以及这些改变对认知/临床完整性的影响。将神经递质受体密度与多模态神经成像相结合的类似多尺度大脑模型也在开发中。与 MCM 方法类似,计划在未来的软件版本中纳入空间分子信息和外部输入效应估计。
我们共享NeuroPM-box的目标有三个:(i)加快研究和临床使用:(ii)就需要解决的任何限制和转化差距寻求反馈:(iii)进一步验证NeuroPM-box,从而提高其适用性。必须强调,虽然本文中提出的 cTI 结果基于特定数据类型(转录、组织病理学数据),但可用于此技术的数据模式的种类和数量没有限制。单细胞转录分析与当前 cTI 实现同样可行,有可能与提出的几个轨迹推理方法进行直接比较。此外,在补充分析中,我们正在研究 cTI 同时集成不同数据模式(分子、多模态神经成像和/或若干认知/行为/临床评估)的能力,这将是我们下一步神经退化研究的主要重点。
为了在使用大规模数据集时进行质量控制,NeuroPM-box目前包括特定的离群值检测方法,以及通过插补进行数据校正。应用先进的神经科学计算技术时,一个共同关心的问题是运行时间效率。大多数NeuroPM-box的优化算法已经实施,以尽量减少计算时间。例如,cTI可以在几分钟内分析数千个受试者和大规模的组学数据。但是,基于微分方程的方法(ESM,MCM)在计算上成本更高。与其他可用技术(如信任区域反射算法)相比,这些方法的优化可以显著缩短计算时间,同时不影响准确性。但是,常规工作站分析数百个具有多模式纵向成像数据的受试者可能需要几天时间(具体取决于模式、大脑区域和时间点的数量)。我们正计划将软件上传到流行的高性能计算(HPC)门户网站,如神经科学网关(NSG,http://www.nsgportal.org)和 CBRAIN(http://www.cbrain.ca)。最后,为了提高软件的通用性,我们还致力于将数据输入扩展到流行的组织格式,包括脑成像数据结构(BIDS)标准。















 9266
9266











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









