







 本文介绍了一种新的方法,ZINB-BasedGraphEmbeddingAutoencoder(scTAG),用于单细胞RNA测序数据的分析和细胞类型注释。通过结合零膨胀负二项模型和图卷积神经网络,scTAG能有效处理数据的稀疏性和高异质性,提供更精确的细胞群体理解和功能解析。
本文介绍了一种新的方法,ZINB-BasedGraphEmbeddingAutoencoder(scTAG),用于单细胞RNA测序数据的分析和细胞类型注释。通过结合零膨胀负二项模型和图卷积神经网络,scTAG能有效处理数据的稀疏性和高异质性,提供更精确的细胞群体理解和功能解析。
Yu, Z., Lu, Y., Wang, Y., Tang, F., Wong, K.-C., & Li, X. (2022). ZINB-Based Graph Embedding Autoencoder for Single-Cell RNA-Seq Interpretations. Proceedings of the AAAI Conference on Artificial Intelligence, 36(4), 4671-4679. https://doi.org/10.1609/aaai.v36i4.20392
科普:
单细胞RNA测序(scRNA-seq)是一种高度精细的技术,用于分析单个细胞水平上的基因表达。它能够揭示细胞群体内部的多样性和功能差异,从而提供了对细胞类型、亚型和状态的更深入理解。
在单细胞RNA测序中,单个细胞的RNA被提取并转录成cDNA,然后通过高通量测序技术进行测序。这种方法可以识别并量化每个单个细胞中的RNA转录本,从而揭示不同细胞之间的基因表达差异。
单细胞RNA测序已经在许多生物学领域取得了重要的应用,包括发育生物学、免疫学、肿瘤学等。它可以帮助科学家们理解细胞的功能和互作,发现新的细胞类型,并揭示疾病发生和发展的机制。
当我们谈论单细胞RNA测序时,我们在研究基因表达的个体细胞水平。这与传统的RNA测序方法有很大的不同,传统方法会混合来自许多细胞的RNA,从而掩盖了细胞间的差异。
单细胞RNA测序的过程可以简单地描述为以下几个步骤:
1. **单细胞样品的准备**:首先,从样品中收集单个细胞。这可能需要使用细胞分选技术,如流式细胞术或微流控技术,以分离单个细胞。
2. **RNA的提取和反转录**:从单个细胞中提取RNA,并将RNA反转录成相应的cDNA。这一步通常涉及到使用逆转录酶将RNA转录成cDNA。在这一过程中,一些测序样本可能需要额外的预处理步骤,如对RNA进行放大。
3. **文库制备**:将cDNA片段转化为文库,通常是通过添加适配器序列来实现。文库制备过程中的每个细胞都会被加上一个唯一的分子标识符(UMI),以区分同一细胞内不同RNA分子的数量。
4. **高通量测序**:将文库进行高通量测序,通常使用Illumina或其他类似平台。在测序过程中,记录每个RNA分子的序列。
5. **数据分析**:对测序数据进行分析,这包括了预处理、细胞识别、表达量估计、细胞类型鉴定等步骤。在这一阶段,可以使用各种算法和工具,如Seurat、Scanpy等,来处理和分析数据。
通过单细胞RNA测序,我们可以识别和比较细胞群体中的不同细胞类型,揭示细胞间的转录差异,发现新的亚型和状态,以及探索细胞在发育、疾病和其他生物学过程中的功能。这项技术已经成为生命科学领域的重要工具,为我们提供了深入了解细胞的机会。
注:在生物学和生物技术领域,高通量通常指的是能够以高效率和大规模地进行实验或测量的技术。在单细胞RNA测序中,高通量测序是指能够以高通量方式快速测序大量细胞中的RNA。这种高通量的测序技术通常使用Illumina等平台,能够同时测序数百万到数十亿个DNA片段或RNA分子,从而大大提高了测序效率和覆盖度。高通量测序技术的优势在于它能够在较短的时间内生成大量的数据,从而使得对生物样品的深入分析和理解成为可能。在单细胞RNA测序中,高通量测序使得我们能够同时测序大量的单个细胞,并对它们的基因表达进行详尽的分析,揭示细胞群体内的多样性和功能差异。
细胞类型注释指的是将单细胞RNA测序数据中的细胞进行分类和标记,以识别它们所属的细胞类型或亚型的过程。在单细胞RNA测序数据中,每个细胞的基因表达模式都是独特的,因此可以根据这些模式将细胞分为不同的类型或亚型。
细胞类型注释的目标是确定细胞的身份,并将其归类为已知的细胞类型,例如神经元、免疫细胞、肌肉细胞等。这对于理解组织、器官或整个生物系统中细胞的功能和相互作用至关重要。
常见的细胞类型注释方法包括聚类分析、细胞标记物鉴定、细胞特异基因表达分析等。聚类分析是最常用的方法之一,通过对细胞基因表达数据进行聚类,将具有相似表达模式的细胞分到同一类别。细胞标记物鉴定则是通过识别与特定细胞类型相关的基因或蛋白质标记来确定细胞类型。而细胞特异基因表达分析则是通过寻找在某一细胞类型中高度表达的基因来推断细胞类型。
细胞类型注释是单细胞RNA测序数据分析的关键步骤之一,它为我们理解细胞组成和功能提供了基础,并为进一步的生物学研究提供了重要的信息。
摘要
单细胞RNA测序(scRNA-seq)可以在单细胞水平上提供基因组范围内的基因表达水平的高通量信息,从而精确了解单个细胞的转录组信息。不幸的是,快速增长的单细胞RNA测序数据以及普遍存在的细胞丢失事件给细胞类型注释带来了巨大的挑战。本文提出一种基于单细胞模型的深度图嵌入聚类(scTAG)方法,它基于深度图卷积神经网络同时学习细胞间拓扑结构表示并识别细胞簇。scTAG将零膨胀负二项(ZINB)模型集成到拓扑自适应图卷积自动编码器中,以学习低维潜表示,并采用Kullback-Leibler(KL)散度进行聚类任务。通过同时优化聚类损失、ZINB损失和细胞图重构损失,scTAG以端到端的方式同时优化簇标签分配和特征学习,并保留拓扑结构。
一、论文动机
单细胞RNA测序(scRNA-seq)技术使得揭示单个细胞的遗传异质性成为可能,这对于基于转录组特征来表征细胞类型(Kolodziejczyk等人,2015年)、研究发育生物学(Chowdhury,2021年)、发现复杂疾病(Costa等人,2013年)以及推断细胞轨迹(Tran和Bader,2020年)至关重要。因此,准确鉴定细胞类型已成为单细胞RNA测序分析的关键步骤(Macosko等人,2015年)。聚类已被证明是细胞类型注释最有效的方法,因为它可以以无偏见的方式识别细胞类型(Kiselev、Andrews和Hemberg,2019年)。在早期研究中,传统的聚类方法,如K均值(MacQueen等人,1967年)、层次聚类(Johnson,1967年)和基于密度的聚类(Kriegel等人,2011年),已被应用于解决聚类任务。然而,对单细胞RNA测序数据进行聚类分析仍然是一项统计学和计算上的挑战,这是因为基因组覆盖度的高度异质性以及一些技术限制导致单细胞RNA测序数据非常稀疏,并且具有大量的零元素(Angerer等人,2017年;Grün,Kester和Van Oudenaarden,2014年)。因此,开发有效的计算方法以释放单细胞RNA测序的全部潜力是至关重要的。
已经开发了几种聚类方法来解决这些限制。大多数研究采用了复杂的技术,涉及迭代聚类,例如,CIDR是一种基于PCA的快速算法,用于基于不相似矩阵的插补和聚类(Lin、Troup和Ho,2017年)。SC3提出了一种单细胞RNA测序数据的共识聚类框架,使用PCA和拉普拉斯变换降低基因维度(Kiselev等人,2017年)。SIMLR使用多核学习来找到更稳健的距离度量,并解决高水平的丢失事件(Wang等人,2017年)。然而,由于基因表达水平的极端稀疏性,这些计算方法通常倾向于在单细胞RNA测序数据上提供次优的结果。此外,大多数方法依赖于完整的图拉普拉斯矩阵,这会带来高计算和存储成本。
近年来,已成功发展了深度嵌入式聚类方法,用于建模高维稀疏的单细胞RNA测序数据;例如,scDeepcluster(Tian等人,2019年)、scDCC(Tian等人,2021年)、scziDesk(Chen等人,2020年)、scDHA(Tran等人,2021年)和DCA(Eraslan等人,2019年)。它们可以通过学习高度自信的分配,使用辅助目标分布来迭代地改进聚类,从而实现更好的聚类结果。然而,这些深度嵌入式聚类方法往往忽视了结构信息传播和节点关系。最近,新兴的图神经网络(GNNs)已被证明能够自然地捕捉通过邻居信息传播的图结构信息(Zeng等人,2020年)。图嵌入聚类通常结合了深度自动编码器和图聚类算法,可以学习潜在的紧凑表示,以探索丰富的内容和结构信息(Nie、Zhu和Li,2017年)。
受上述观察的启发,我们在此提出了一种基于单细胞模型的深度图嵌入聚类方法,名为scTAG,它同时从自动编码器中学习细胞间拓扑表示并识别细胞簇(Du等人,2017年)。首先,我们利用零膨胀负二项模型(ZINB)来捕捉数据的全局概率结构,通过学习包括均值、离散度和丢失概率在内的三个特征分布参数。然后,scTAG提出了一种基于ZINB的图卷积自动编码器,以在低维潜在空间中保留细胞的拓扑结构。之后,利用Kullback–Leibler(KL)散度来优化聚类过程。最后,scTAG可以结合三种训练损失,包括聚类损失、ZINB损失和细胞图重构损失,来优化细胞簇标签分配并学习细胞间拓扑表示,从而产生优越的聚类结果。
二、贡献
- 提出了一种基于单细胞模型的深度图嵌入聚类方法,称为scTAG,它将零膨胀负二项模型整合到拓扑自适应图卷积自动编码器中,以捕捉数据的全局概率结构。
- scTAG构建了一个细胞图,并使用拓扑自适应图卷积自动编码器来保留单细胞RNA测序数据中的拓扑结构信息和细胞间关系。
- 第一篇将ZINB整合到图卷积自动编码器中,以建模高度稀疏和过分离的单细胞RNA测序数据的文章。
- 在16个真实单细胞RNA测序数据集上评估了我们的模型和最先进的竞争方法。结果表明,scTAG优于所有基线方法。
三、方法
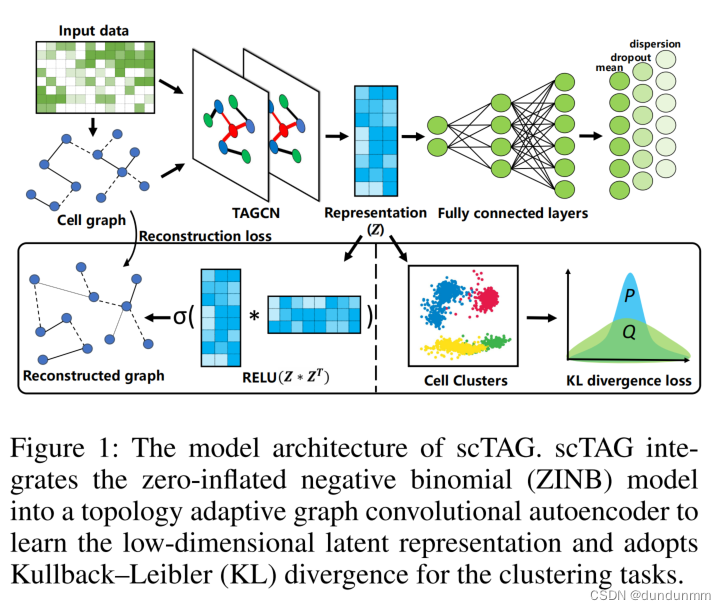
1、数据预处理
论文以scRNA-seq基因表达矩阵X作为输入,其中Xij表示第i个细胞(1 ≤ i ≤ N)中第j个基因(1 ≤ j ≤ O)的表达个数。
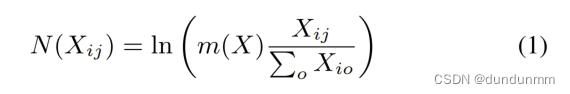
其中,m(X)代表细胞总表达值的中位数。根据公式(1),离散数据被平滑处理,并通过自然对数变换重新调整。标准化后,根据由scanpy包(Wolf, Angerer, and Theis 2018)计算的标准化离散值的排名,选择了前t个高变异基因,以识别具有高水平信息的基因。
2、细胞图
在本研究中,利用从图自编码器学习到的嵌入来保留细胞之间的关系和邻居信息。与以前的研究类似(Wang et al. 2021),采用KNN算法来构建细胞图,图中的每个节点代表一个细胞。实际上,如果存在节点a和b,并且在a和b之间存在一条边;如果a是b的邻居,且在k个最短距离内,其中k被设定为15。欧氏距离被计算来描述节点之间的关联,以发现k个最短距离。之后,构建的细胞图是一个无向图,边的权重统一设置为1。
3、拓扑自适应图卷积自编码器
为了捕获图结构和节点关系,本文开发了一种变种的图卷积自编码器,使用拓扑自适应图卷积网络(TAGCN)(Du et al. 2017)作为图编码器。其思想是TAGCN在每一层使用K个图卷积核来提取不同尺寸的局部特征,从而避免了近似卷积核不能充分提取图信息的缺点,从而增强了模型对scRNA-seq数据的学习能力。
基因表达矩阵X和标准化邻接矩阵A(上一节中计算的细胞图)被用作输入。
考虑到第l个隐藏层,假设每个节点在特征映射后具有Cl个特征,图卷积过程可以定义如下:
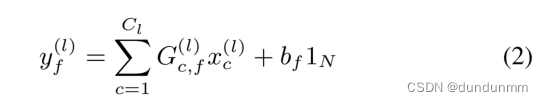
其中,表示第f个输出特征图;bf 是可学习的偏置;
表示TAGCN中的多项式卷积核,其内部结构使用K个图卷积核来提取不同尺寸的局部特征,定义如下:
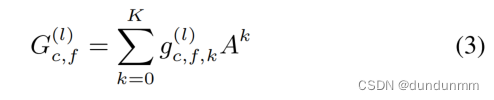
其中, 表示多项式系数。采用标准化邻接矩阵A可以实现整个卷积操作的更稳定计算。在每次图卷积操作之后,对输出应用非线性操作,如下所定义:
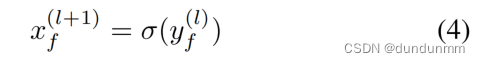
由于大部分的结构和信息都通过TAGCN编码器保留在scRNA-seq数据X的潜在嵌入表示Z中,因此图自编码器的解码器可以被定义为潜在嵌入之间的内积:
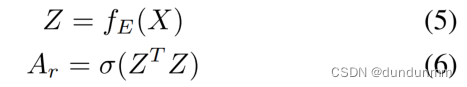
其中,fE表示TAGCN编码器函数;Ar是重构的邻接矩阵。因此,在学习过程中,应该最小化A和Ar的重构损失,如下所示:
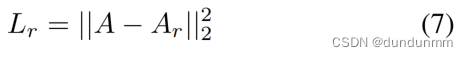
4、基于ZINB的图卷积自编码器
为了更好地从潜在嵌入表示Z解码以捕获单细胞RNA测序数据的结构,本文将零膨胀负二项式(ZINB)模型集成到拓扑自适应图卷积自编码器中,以捕获数据的全局概率结构。首先分析了在先前的研究中使用零膨胀负二项式分布(ZINB)近似表示单细胞RNA测序数据分布的原因(Risso et al. 2018; Miao et al. 2018)。
定理1 单细胞RNA测序基因表达计数矩阵的数据分布可以近似为零膨胀负二项分布(ZINB)。
证明。单细胞RNA测序基因表达计数矩阵的数据分布通常符合三个特征:1)离散性;2)方差大于均值;3)包含许多零值,包括非表达基因(真零)或由于技术原因(dropout零)。接下来,证明ZINB分布可以模拟这三个属性。ZINB定义如下:
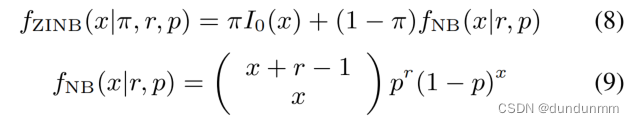
其中,π表示零值的比例;I0(x)是一个指示函数,当x = 0时等于1,否则等于0;r和p是负二项分布(NB)的参数,分别表示成功次数和概率。由于NB分布属于离散分布,因此ZINB也满足离散分布的性质。当x = 0时,ZINB可以通过π预测dropout零的概率(dropout率),推导如下:
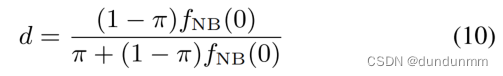
同时,可以证明方差大于均值,遵循NB分布。假设均值为E(x),定义如下: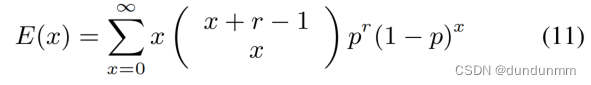
令 x' = x - 1 且 r' = r + 1 ,则有: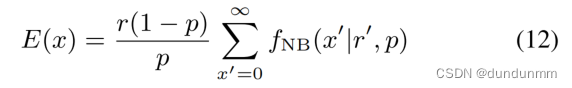
由于NB是离散分布,所有概率之和等于1;即
![]()
因此,![]()
假设方差为Var(x),定义如下: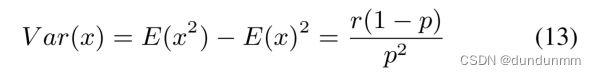
然后,可以得到E(x)和Var(x)之间的关系: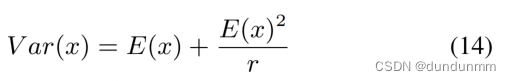
由于 r > 0 ,所以 Var(x) > E(x) 。
在此基础上,论文应用ZINB分布模型来模拟数据分布,以捕获scRNA-seq数据的特征。然后,基于ZINB的图卷积自编码器代替常规图自编码器,该模型训练以尝试重构其输入,定义如下:
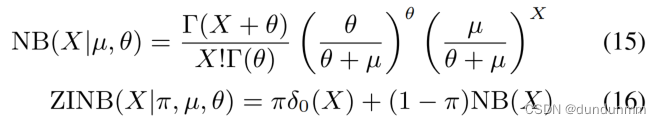
其中,μ和θ分别表示均值和离散度;π是零点质量的权重。比例 θ / (θ + μ) 替换了方程(9)中的概率 p。之后,用三个全连接层来估计潜在嵌入表示 Z 中的参数 {π, μ, θ},如下所示:
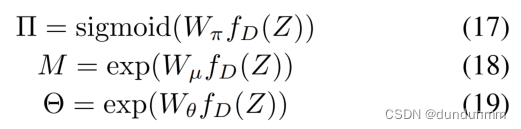
其中,fD 是一个具有128、256和512个节点的三层全连接神经网络;W 表示损失函数的学习权重;Π、M 和 Θ 都是参数矩阵,分别表示网络输出的信息丢失概率、均值和离散度。激活函数的选择取决于参数的范围和定义。信息丢失概率介于 0 和 1 之间,因此选择了 sigmoid 函数。此外,由于均值和离散度的非负值,应用了指数函数。ZINB 分布的负对数似然可以作为原始数据 X 的重构损失函数,可以定义如下:
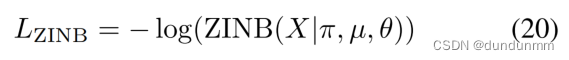
5、自优化深度图嵌入聚类
由于深度图嵌入聚类方法是无监督的,并且在训练过程中没有标签的指导,因此无法获得良好的优化反馈。因此,论文设计了一种自优化嵌入算法,将潜在嵌入输入到自优化聚类模块中。
与DEC相同,应用了KL散度和学生t-分布:
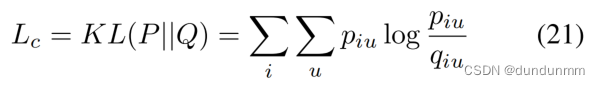
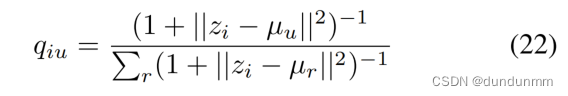
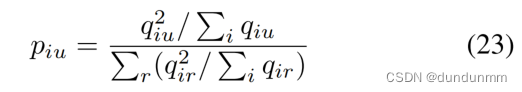
在整个训练过程中,图自编码器嵌入和聚类学习是共同优化的。最小化以下总目标函数:
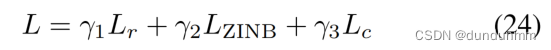
四、实验
1、实验数据
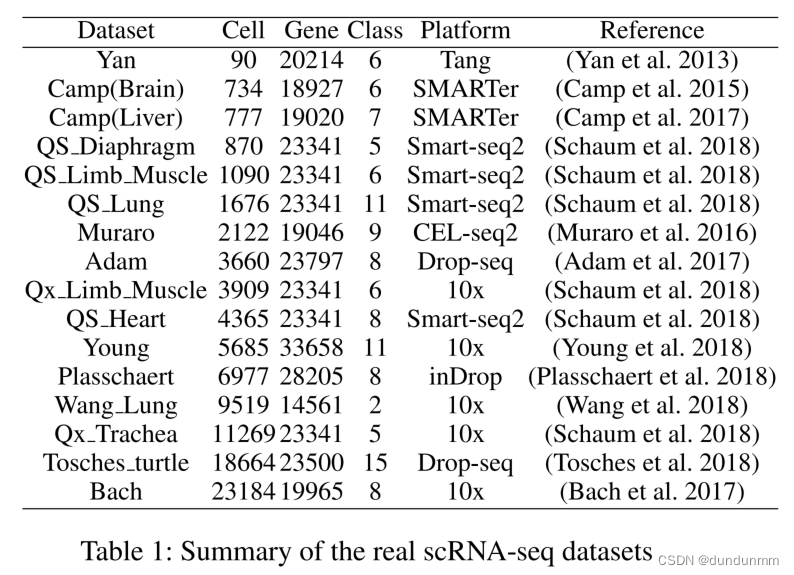
论文将所提模型的性能与其他基线方法在来自几个代表性测序平台的 16 个真实世界单细胞RNA测序数据集上进行了比较。实验中使用的这 16 个单细胞RNA测序数据集来自最近发表的关于单细胞RNA测序实验的论文,详细信息见表1。所有这 16 个数据集来自不同的物种,包括小鼠和人类,以及来自不同的器官,如大脑、肺和肾。具体而言,细胞数量范围从 90 到 23184,基因数量范围从 14561 到 33658。
2、实验结果
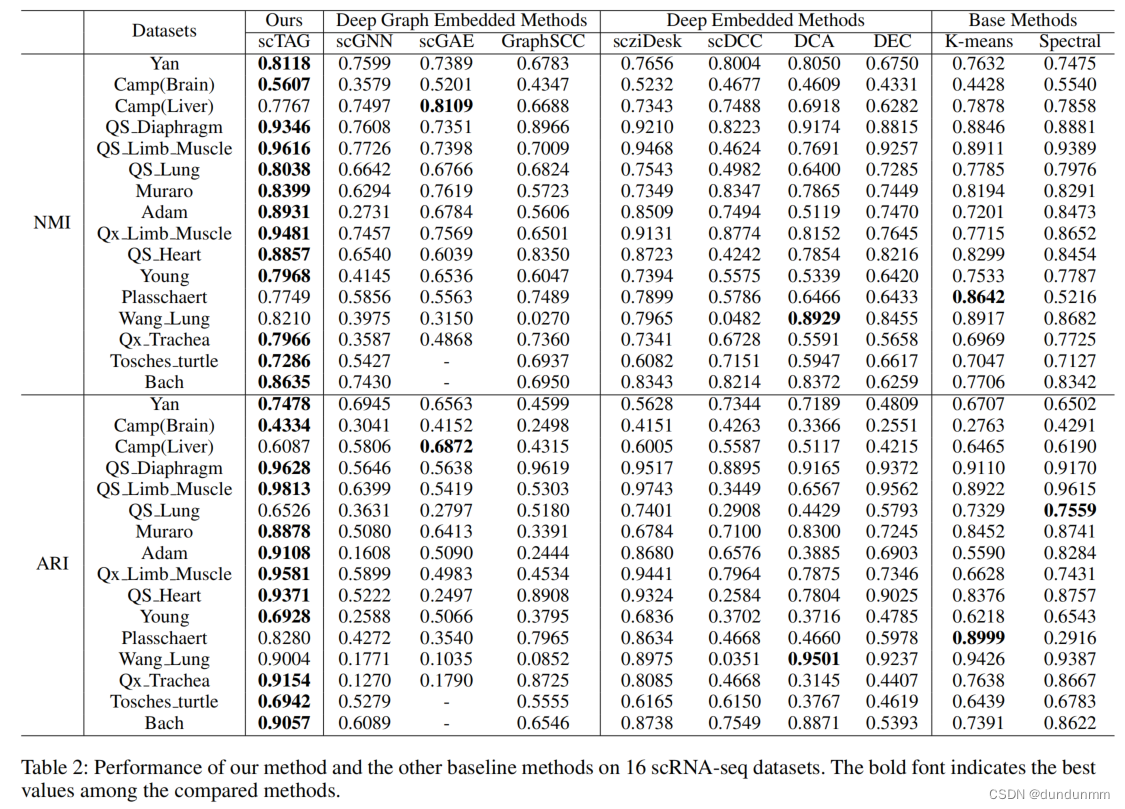
论文很好的将深度聚类模型与单细胞RNA测序工作进行了结合,重点在于定理ZINB分布与单细胞RNA测序基因表达计数矩阵的数据分布近似一致,从而使得学习出来的潜在分布符合基因表达规律。















 2543
2543

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









