







单细胞转录组 —— 原始数据处理实战(simpleaf)
前言
Alevin-fry 是一个快速、准确且内存节约的单细胞和单核数据处理工具。
Simpleaf 是用 Rust 编写的程序,它提供了一个统一且简化的界面,用于通过 alevin-fry 流程处理一些最常见的单细胞数据。
准备
开始之前,我们先在终端创建一个 conda 环境,并安装所需的软件包。
Simpleaf 依赖于 alevin-fry、salmon 和 pyroe。它们都可以在 bioconda 上找到,并会在安装 simpleaf 时自动安装。
安装虚拟环境和软件
conda create -n af -y -c bioconda simpleaf
conda activate af
simpleaf 需要环境变量 ALEVIN_FRY_HOME 来存储配置和数据
export ALEVIN_FRY_HOME=$(dirname $(which simpleaf))
使用 set-paths 命令查找所需工具的路径,并在 ALEVIN_FRY_HOME 文件夹中写入一个配置 JSON 文件
simpleaf set-paths
构建索引
索引命令有两种输入形式:
- 将参考基因组
FASTA和GTF作为输入,然后使用roers(自带的工具,无需单独安装)spliced+intronic(splici)参考或spliced+unspliced(spliceu)参考 - 将单个参考序列文件(即
FASTA)作为输入(直接参考模式)。
在扩展参考的模式下,构建完成扩展的参考之后,将使用 piscem build 或 salmon index 命令(可以自选)对生成的参考进行索引,并在索引目录中存储一个包含 3 列的转录本到基因的文件(t2g_3col.tsv),以供后续使用。
输出目录将包含 ref 和 index 子目录,前者包含从提供的基因组和 GTF 中提取的 splici 参考,后者包含在此参考基础上构建的索引。
在直接参考的模式下,提供的 fasta 文件(使用 ——ref-seq 参数)将直接提供给 piscem 或 salmon 来构建索引。输出目录将包含一个索引子目录,其中包含基于此参考文件构建的索引。
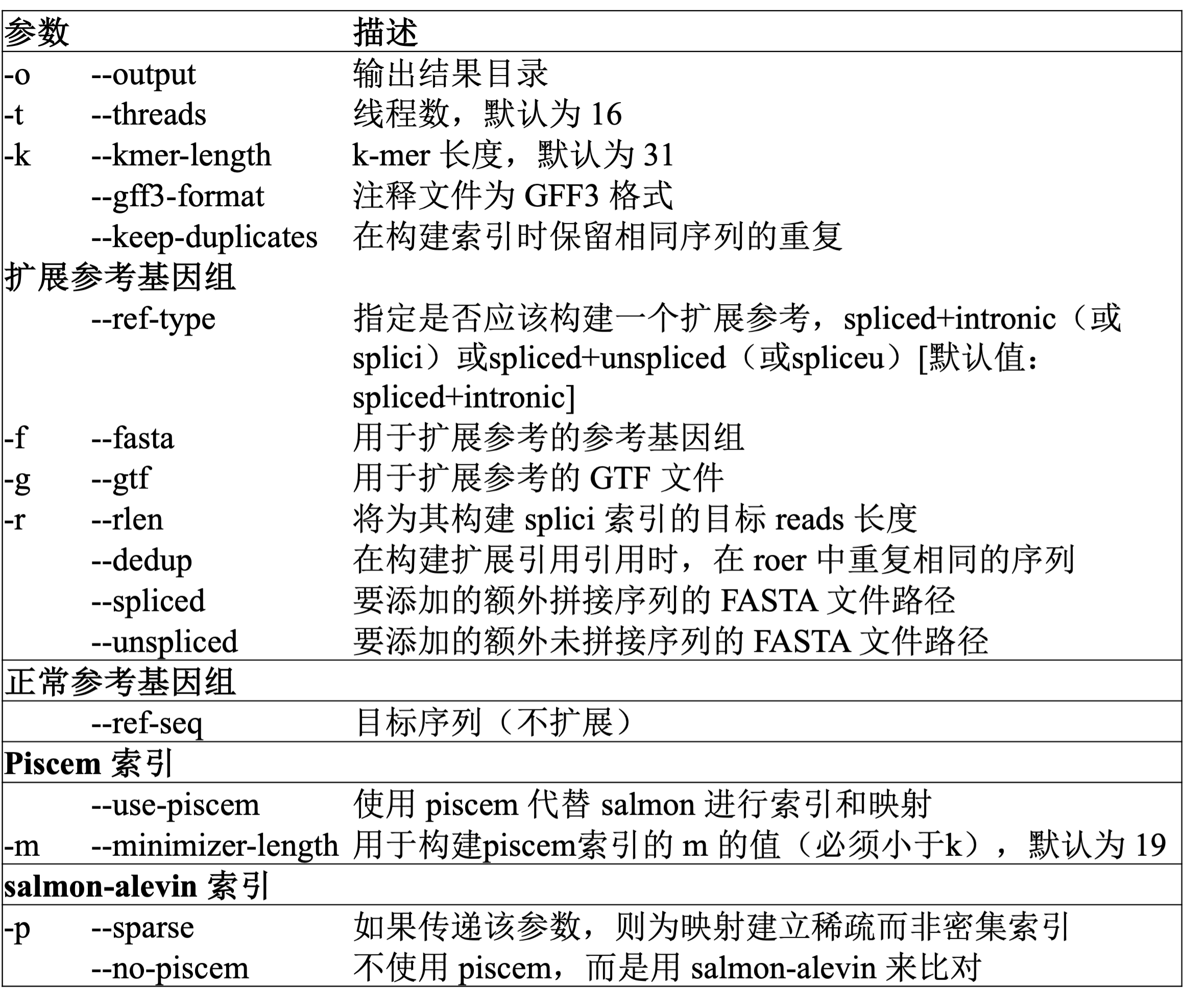
具体参数可以参考如下
下载参考基因组信息,例如
wget http://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
wget http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.primary_assembly.annotation.gtf.gz
可以用我们之前已经下载过的,不要重复下载。
注意:simpleaf 要求 FASTA 文件中的碱基都为大写,如果你下载的版本不是,可以使用 seqkit 工具进行装换。例如
conda install -c bioconda seqkit
seqkit seq -u genome/hg19.fa > genome/hg19_upper.fa
构建参考基因组的索引,默认使用 piscem 构建索引,但是后面在比对时容易出现问题,所以使用 salmon 来比对。
使用 --no-piscem 来禁用 piscem
simpleaf index \
-o simpleaf_hg19 \
-f /path/to/genome/hg19.fa \
-g /path/to/annotation/hg19.refGene.gtf \
-r 90 --no-piscem \
-t 8
在输出目录 simpleaf_hg19 中,ref 文件夹包含 splici 参考;index 文件夹包含基于 splici 参考构建的 salmon 索引。
表达定量
使用 quant 可以进行细胞条形码校正、UMI 解析并生成计数矩阵。其输入可以是
- 索引、reads 和其他实验信息,如
UMI和barcode位置 - 包含比对结果和有关实验信息的目录
然后运行 alevin-fry 流程的所有相关步骤。
从头开始处理一个新数据集时,你应该选择第一种方式(需要提供 --index、--reads1 和 --reads2 参数)。如果 --reads1 和 --reads2 参数有多个文件,必须用逗号(,)分隔。
另一方面,如果您已经进行了定量分析,或者由于其他原因已经对读数进行了比对,并生成了 RAD 文件,则可以使用 --map-dir 参数直接从比对结果启动后续分析。
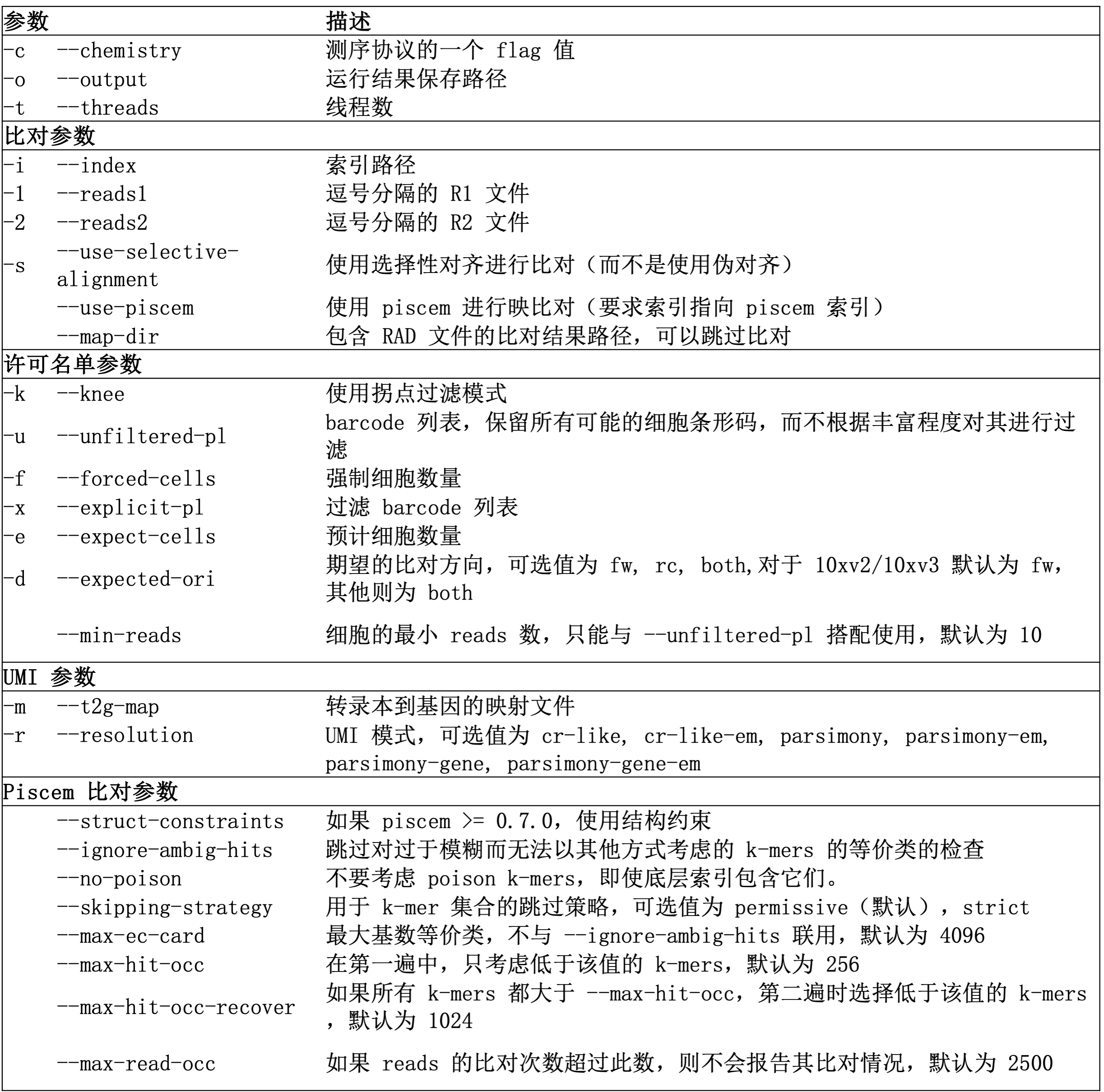
后一种方法便于测试不同的定量方法(例如不同的过滤选项或 UMI 解析策略)。主要的参数包括
关于 --chemistry 参数
--chemistry 选项可以是描述特定化学成分的字符串,也可以是描述条形码几何形状、UMI 和可比对读取的字符串。
例如,字符串 10xv2 和 10xv3 将分别应用 10x chromium v2 和 v3 协议的适当设置。
不过,如果您要使用的不是程序内预注册选项,也可以提供一般形式,用于表述序列状态。例如 10xv2 可以表示为
1{b[16]u[10]x:}2{r:}
其中 1 表示 read1 文件,花括号内的参数用于描述 read1, b16 表示 1-16 位的碱基是 barcode 序列,u[10] 表示第 17-26 位的碱基是 UMI 序列,x: 表示后面的所有序列将会被丢弃;
类似地,2 则表示 read2 文件,r: 表示整条序列都是 cDNA 序列。
所以 10xv3 也可以表示为 "1{b[16]u[12]x:}2{r:}"。
序列标识有可能出现重复,在这种情况下,它们将被提取并连接在一起。例如,1{b[16]u[12]b[4]x:} 表示我们应提取 1-16 位和 29-32 位的碱基,并将它们连接起来以获得完整的条形码。
你可以将经常使用的几种自定义字符添加到 ALEVIN_FRY_HOME 目录中的 custom_chemistries.json 文件中。
例如,将下面的内容放入该文件后,您就可以向 simpleaf quant 命令传递 --chemistry flarb,它就会将 reads 解释为具有指定的形式。
{
"flarb" : "1{b[16]u[12]x:}2{r:}"
}
实战
10x scRNA-seq
我们从 10x Genomics 公司下载的人类脑肿瘤数据集 Brain_Tumor_3p_LT_fastqs。
wget https://cf.10xgenomics.com/samples/cell-exp/6.0.0/Brain_Tumor_3p_LT/Brain_Tumor_3p_LT_fastqs.tar
解压数据
tar -xvf Brain_Tumor_3p_LT_fastqs.tar
表达定量分析
# 搜索 FASTQ 文件,并使用 , 连接多个文件
reads1="$(find -L Brain_Tumor_3p_LT_fastqs -name "*_R1_*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd,)"
reads2="$(find -L Brain_Tumor_3p_LT_fastqs -name "*_R2_*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd,)"
simpleaf quant \
-c 10xv3 -t 8 \
-1 $reads1 -2 $reads2\
-i simpleaf_hg19/index \
-u 3M-february-2018.txt \ # 10xv3 barcode
-r cr-like \
-m simpleaf_hg19/index/t2g_3col.tsv \
-o simpleaf_brain
运行这些命令后,可在 ·simpleaf_brain/af_quant/alevin 文件夹中找到定量信息。
该目录下有三个文件:
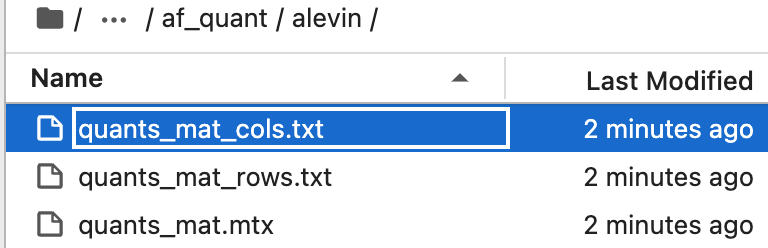
quants_mat.mtx:计数矩阵quants_mat_cols.txt:矩阵每列的基因名称quants_mat_rows.txt,矩阵每行经过校正、过滤的细胞条形码
值得注意的是,alevin-fry 是在 USA-mode(未剪接变体(U)、剪接变体(S)和剪接模糊变体(A))下运行的,会对每个基因的剪接和未剪接状态都进行定量分析,生成的 quants_mat_cols.txt 文件的行数等于注释基因数的 3 倍
Drop-seq
与 kb-python 教程一样,我们使用的数据集是 GSE178612,用于研究 FoxM1 与 Rb 基因在小鼠乳腺癌中的相互作用。
我们选择其中一个样本 GSM5394388,下载其原始数据
prefetch SRR14872449
解压
fastq-dump SRR14872449/SRR14872449.sra --split-files --gzip -O SRR14872449
构建小鼠的索引
simpleaf index \
-o simpleaf_mm10 \
-f /path/to/genome/mm10.fa \
-g /path/to/annotation/mm10.refGene.gtf \
-r 90 --no-piscem \
-t 8
定量分析,使用 -k 参数,利用拐点来矫正 barcode,禁用 piscem
simpleaf quant \
-c "1{b[12]u[8]x:}2{r:}" -t 8 \
-1 SRR14872449/SRR14872449_1.fastq.gz \
-2 SRR14872449/SRR14872449_2.fastq.gz \
-i simpleaf_mm10/index \
# -u 3M-february-2018.txt \ 不需要
-r cr-like -k --no-piscem \
-m simpleaf_mm10/index/t2g_3col.tsv \
-o simpleaf_drop
Indrop
与 kb-python 教程一样,使用包含 6 例原发性胰腺癌组织的单细胞 RNA 测序和空间转录组学 GSE111672 数据集
我们随便选择一个样本 GSM3036909 下载原始数据
prefetch SRR6825055
解压
fastq-dump SRR6825055/SRR6825055.sra --split-files --gzip -O SRR6825055
表达定量,其中 barcode 需要自己下载合并,barcode 和 UMI 组成需要自己构建字符串
simpleaf quant \
-c "1{b[12]u[8]x:}2{r:}" -t 8 \
-1 SRR6825055/SRR6825055_1.fastq.gz \
-2 SRR6825055/SRR6825055_2.fastq.gz \
-i simpleaf_hg19/index \
# -u indrop_barcode.txt \
-r cr-like -k --no-piscem \
-m simpleaf_hg19/index/t2g_3col.tsv \
-o simpleaf_indrop
因为 indrop 的 gel_barcode1_list.txt 文件长度不一,会有问题。所以在这里我们使用 -k 参数,利用拐点的方式矫正 barcode。
读取结果
Python
我们可以使用 pyroe 中的 load_fry 函数将计数矩阵作为 AnnData 对象加载到 Python 中。
import pyroe
quant_dir = 'simpleaf_brain/af_quant'
adata_sa = pyroe.load_fry(quant_dir)
默认情况下,Anndata 对象的 X 层加载为每个基因的剪接计数和模糊计数之和。不过,最近的工作Pool et.al, 2022 研究表明,即使在 scRNA-seq 数据中加入内含子计数,也可能会提高灵敏度并有利于下游分析。
虽然利用这一信息的最佳方法仍在研究之中,但由于 alevin-fry 会自动量化每个样本中的剪接、非剪接和模糊读数,因此包含每个基因总计数的计数矩阵可通过以下方式简单获得:
import pyroe
quant_dir = 'simpleaf_brain/af_quant'
adata_usa = pyroe.load_fry(quant_dir, output_format={'X' : ['U','S','A']})
R
在 R 中,我们可以使用 fishpond 包中类似的 loadFry 函数。
首先,安装包
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("fishpond")
导入包并读取数据
library(fishpond)
loadFry(fryDir = "simpleaf_brain/af_quant")
















 1219
1219

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











