










摘要:在单原子催化剂(SAC)中,支撑锚定位点的复杂性创造了具有不同配位环境的大量单原子物种。迄今为止,给定 SAC 中这些不同单原子物种的数量分布仍然难以捉摸。最近,CeO2负载的金属SAC通过多种合成策略调节其局部环境而得到了广泛的研究。然而,由于缺乏定量描述,阐明位点特异性反应性并调节其转化仍然具有挑战性。在这里,我们证明了两种不同的 Pt/CeO2SAC 可以通过氧化和非氧化分散体可逆地生成,尽管 Pt 电荷态和配位数相似,但它们含有不同的 Pt1On–Ceδ+单原子物种。通过拉曼光谱和计算研究,我们半定量地揭示了每个特定 SAC 中不同 Pt1On –Ceδ+物种的分布。值得注意的是,仅占14.2%的少数物质Pt1O4 –Ce3+–O v在其他丰富的对应物(即Pt1O4 –Ce4+和 Pt1O4 –Ce4+)中提供了最高的低温CO氧化反应活性。第二个最近的氧空位(Ov)不仅与附近的活性金属位点协同作用以降低反应势垒,而且还促进循环非氧化和氧化分散过程中从六配位位点到四配位位点的动态转变。这项工作阐明了给定 SAC 中各种单原子物种的定量分布和动态转变,为描述 SAC 的不均匀性提供了更内在的描述符和定量测量。
Introduction
单原子催化剂(SACs)因其最大化原子利用率和特异性反应性而引起巨大兴趣。类似于均相催化剂,SACs的单原子特性通常相较于其他非均相催化剂表现出较高的选择性。然而,由于载体上存在大量不同的锚定位点,包括第一/二壳协同结构和表面次协同位点(缺陷、转角或台阶边缘),SACs在现实中不仅仅是均相的。这些锚定位点强烈调控单原子中心的局部环境,从而影响其位点特异性反应性。尽管表面锚定位点的复杂性,迄今为止,仅采用了平均描述符,如电荷状态和配位数(CN),来描述SACs的整体状态,这并不足以反映SACs的多样性和不均匀性。迄今为止,对于一个给定的SAC中各种单原子物种的定量分布仍然是难以捉摸的。
CeO2载体上支持的金属单原子催化剂(例如Pt,Pd)最近在低温CO氧化作为一种探针反应中受到了密切关注。已经报道了多种多样的单原子催化剂,其电荷状态、配位和表面位点各异,导致了反应活性的显著差异。然而,由于缺乏定量描述,各种单原子催化剂物种的位点特异性反应仍在争论之中,且缺乏有效途径调控它们的分布。
在这项工作中,通过非氧化(在N2中)和氧化分散(在O2中),可逆地制备了两种不同的Pt1/CeO2单原子催化剂(Pt/CeO2–N600和Pt/CeO2–O600)。我们使用先进的表征技术,包括环境透射电子显微镜(ETEM)、扫描透射电子显微镜(STEM)、X射线光电子能谱(XPS)和扩展X射线吸收精细结构(EXAFS)光谱,揭示了不同Pt1On–Ceδ+单原子种类及其转化的协同结构。基于定量分析和拉曼光谱的计算模拟,我们在每个具体的单原子催化剂中半定量地揭示了不同Pt1On–Ceδ+单原子种类的分布。低温CO氧化被用作一种探针反应,以展示位点特异性反应活性与单原子种类分布的依赖关系。密度泛函理论(DFT)计算进一步验证了邻近氧空位在促进CO氧化和在循环非氧化和氧化分散期间动态转化Pt1On–Ceδ+物种中的作用。
Results and Discussion
Pt 分散的原位观察
图1a展示了通过非氧化和氧化分散制备Pt/CeO2单原子催化剂的设计方案。它们分别被标记为Pt/CeO2–Ox和−Nx,其中x代表在O2或N2中的预处理温度。原始的Pt/CeO2(1 wt%,图1b)显示了存在小的Pt纳米颗粒(NPs),平均大小为1.6纳米(图S1)。在N2气氛中加热至600°C后观察到Pt NPs的非氧化分散。像差校正的高角度环形暗场扫描透射电镜(AC-HAADF-STEM)成像(图1c)清晰地显示了CeO2表面高度分散的Pt原子。随着分散温度升至800°C,Pt SA物种变得热不稳定,容易聚集成NPs(∼2纳米)(图1d)。相反,在O2气氛中,Pt NPs的氧化分散发生在600°C(图1e),在800°C时仍然保持原子分散而不发生烧结(图1f)。
EXAFS谱学进一步证实了Pt单原子或Pt纳米粒子在CeO2上的分散状态(图1g)。对于Pt/CeO2–N600、Pt/CeO2–O600和Pt/CeO2–O800,仅在∼1.99 Å处识别到Pt–O散射,没有Pt–Pt配位,这证实了Pt物种的原子分散状态。在Pt/CeO2–N800的情况下,Pt–O键的峰值强度大大减弱,而Pt–Pt配位在∼2.72 Å处增强,表明Pt原子聚集成纳米粒子,与STEM观察一致。CO化学吸附的傅里叶变换红外(FTIR)光谱也证实了Pt在Pt/CeO2–N600、Pt/CeO2–O600和Pt/CeO2–O800中的原子分散状态,具有2084和2092 cm-1的特征CO伸展模式,对应于离子型Pt物种上的线性吸附CO。波数上的微小差异表明这些单原子Pt在这些单原子催化剂中具有稍微不同的局部环境。

Figure S3展示了在N2环境中(1mbar,图S3a–c)100到800℃范围内Pt/CeO2催化剂的ETEM图像,随后在O2环境中(1mbar,图S3d–f)进行氧化分散,采用相同的加热程序。随着温度从100升至600℃,Pt NPs的解体发生,颗粒逐渐缩小并逐渐消失。在进一步升温至800 ℃后,Pt NPs重新出现,表明在非氧化气氛下在800℃以上,Pt SAs在CeO2上不稳定。随后,在切换到O2气氛后,最初在600摄氏度观察到氧化分散。由于1 mbarO2下分散动力学相对于1 bar O2的非原位实验(图1)较慢,更高的分散可能需要更长的时间。因此,原位ETEM实验清楚验证了在氧化和非氧化氛围下Pt SAs的动态形成。
识别单原子的配位结构
非氧化和氧化分散的Pt/CeO2 SACs的协同结构通过XPS在Pt 4f(图2a)和Ce 3d核心电平区域(图2b)确定。在Pt 4f核心电平中,可以将Pt0(蓝色区域),Pt2+(粉色区域)和Pt4+(红色区域)的三个主要成分解卷,其中Pt 4f5/2结合能(BE)分别位于71.5、72.9和74.3电子伏特。Ce 3d光谱可以解卷为Ce4+的六个峰(v、v″、v‴、u、u″和u‴分别位于882.48、888.78、898.18、901.13、907.28和916.73电子伏特)和Ce3+的四个峰(v0、v′、u0和u′分别位于879.88、884.88、899.53和902.53电子伏特)。如图2c和表S1总结的,原始的Pt/CeO2催化剂包含约60%的Pt0和约40%的Pt2+物种,它们分别被分配为与CeO2接触的金属Pt NPs和离子界面物种。经氧化分散后,Pt/CeO2–O600和Pt/CeO2–O800均显示出主导的Pt2+组分(87-93%),暗示高度分散的Pt SAs的形成,具有增强的Pt–O键合,与EXAFS和HAADF-STEM结果一致。有趣的是,在N2处理后,在Pt/CeO2–N600中不存在Pt0组分,强烈证明了Pt NPs即使在惰性环境中也发生了类似的再分散过程,导致Pt SAs具有主导的Pt2+状态(约90%)。进一步加热到800°C时,由于Pt SAs变得不稳定,聚集的Pt NPs形成,Pt0组分的小部分重新出现。

通常认为,气态O2对于诱导分散是必要的。然而,根据我们的结果,晶格氧也通过反向氧溢出可能促进了Pt原子的氧化和分散。这一进一步得到证实,即Pt/CeO2–N600的Ce3+/Cetotal值(20.2%)高于Pt/CeO2–O600(15.3%)和Pt/CeO2–O800(13.4%)(图2c和表S1)。在低温下(分别为Pt/CeO2–N600和Pt/CeO2–N800的269和275°C),不存在还原峰,表明在非氧化分散过程中,Ce4+向Pt单原子(SAs)发生反向溢出时,界面处的晶格氧被迅速消耗(图2d)。
X射线吸收光谱(XAS)在Pt L3边缘进一步阐明了Pt/CeO2单原子催化剂(Pt/CeO2–O600和Pt/CeO2–N600)的局部配位环境。图S4中的X射线吸收近边结构(XANES)显示了Pt/CeO2–O600和Pt/CeO2–N600中相似的白线强度,表明由于与表面氧物种的强键合,孤立的Pt物种(Pt2+)具有类似的离子态。通过拟合扩展X射线吸收精细结构(EXAFS,图2c和表S2和S3),进一步分析了局部Pt–O配位数(CNPt–O)。氧化分散的Pt/CeO2–O600和Pt/CeO2–O800单原子催化剂显示略高的CNPt–O,分别为∼4.7和4.5,与非氧化分散的Pt/CeO2–N600单原子催化剂相比(CNPt–O ∼ 4.4)。从XPS和EXAFS的结果可以看出,通过氧化和非氧化分散生成的Pt/CeO2–O600、Pt/CeO2–O800和Pt/CeO2–N600单原子催化剂在平均CNPt–O和电荷状态上具有相似的特征(图2c),但在Ce3+位点和氧空位的数量上存在显著差异。具体而言,Pt/CeO2–O600由Pt1On–Ce4+物种主导,其平均CNPt–O约为∼4.7,而Pt/CeO2–N600包含更多的Pt1On–Ce3+–Ov物种(CNPt–O约为4.4,其中Ov指氧空位),通过从相邻的Ce4+位点发生反向氧溢流形成,导致Ce3+和Ov位点的成对存在(图2e)。密度泛函理论(DFT)计算进一步验证了具有氧空位的Pt1O4–Ce3+–Ov物种对于第二近邻的稳定性(图S5),其比具有氧空位的最近邻物种更稳定,能量差为2.2eV(表S4)。根据上述结果,我们可以证明,像配位数和电荷状态这样的平均描述符不足以反映这两种单原子催化剂中Pt1On–Ceδ+物种的实际变化,尤其是表面Ov位点对于第二近邻缺乏敏感性的Pt配位中心。
Pt单原子的分布和转变
分布和协同转化不同的Pt1On–Ceδ+物种通过拉曼光谱进一步研究(图3a)。所有催化剂显示出CeO2支撑上的强烈带谱在463 cm-1处,这被归因于与立方体[CeO8]单元中Ce–O键的对称伸展相关的F2g带。在Pt/CeO2 SACs上F2g带的上移(图S6)被归因于Pt SA的并入到CeO2晶格中。除了F2g带之外,通过位于∼300、∼520和∼680 cm-1处的带识别了三个额外的拉曼振动模式,这些模式与Pt–O/Pt–O–Ce连接的特征峰有关(图3a)。根据文献,∼300和520 cm-1处的带被归因于Pt–O–Ce连接的非对称振动,而680 cm-1处的带被归因于在CeO2表面上形成的方平面配位的Pt2+离子的Pt–O物种。值得注意的是,在680 cm-1处宽带相邻的693 cm-1处明显可区分出Pt/CeO2–N600和Pt/CeO2–N800样品,这强烈暗示了在同一区域对Pt–O振动有多个组分共存。在∼300、∼520和∼680/693 cm-1处带的峰强度波动表明了给定催化剂中多样的Pt1On–Ceδ+物种的不同分布。
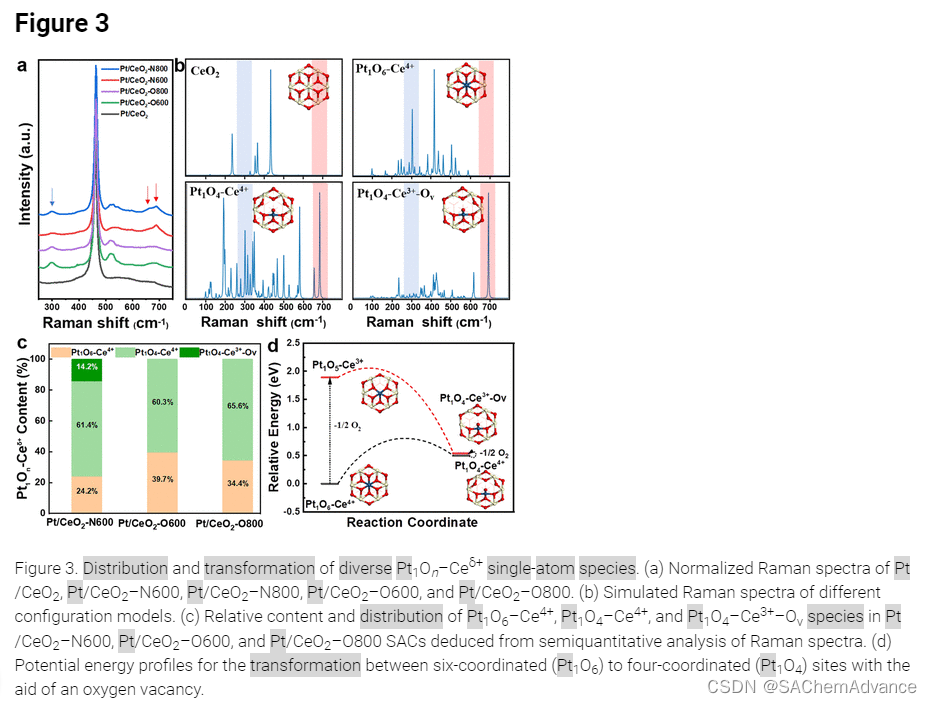
此外,通过声子频率计算和Python program vasp_raman.py,模拟了不同Pt1On–Ceδ+结构的拉曼光谱。构建了包含Pt原子的CeO2(111)或体相CeO2中的19种Pt1On–Ceδ+结构模型,并获得相应的模拟拉曼频率(图S7和表S5)。从19种可能的构型中选择了与实验光谱良好拟合的三种典型Pt1On–Ceδ+物种进行考虑(图3b),它们分别是六配位位点(Pt1O6–Ce4+)、方平面Pt1O4位点(Pt1O4–Ce4+)和带有氧缺陷的方平面位点模型到第二近邻居(Pt1O4–Ce3+–Ov)。通过比较实验光谱(图3a)和模拟光谱中突出显示的特征拉曼带,我们发现方平面几何形状的Pt1O4–Ce4+和Pt1O4–Ce3+–Ov结构都对680/693 cm–1带有贡献,而六配位的Pt1O6–Ce4+主要对∼300和520 cm–1带有贡献(图3b)。
鉴于693 cm–1处尖锐峰与富含氧空位的非氧化分散的单原子Pt(SACs)之间存在强烈的相关性,我们将693 cm–1峰归属为Pt1O4–Ce3+–Ov物种,而680和300 cm–1处的宽带则分别表示Pt1O4–Ce4+和Pt1O6–Ce4+物种。这使得通过分析拉曼峰面积(详见表S6中的计算细节)对这些物种的分布进行半定量分析成为可能。在图S8中展示了以∼299、678和693 cm–1为中心的解卷积拉曼峰,它们分别代表Pt1O6–Ce4+、Pt1O4–Ce4+和Pt1O4–Ce3+–Ov物种。值得注意的是,Pt/CeO2–N600、Pt/CeO2–O600和Pt/CeO2–O800 SACs均来自相同的Pt/CeO2源;因此,我们推断这三种SACs含有相同数量的Pt原子,其中每个Pt1On–Ceδ+的含量总和等于100%。因此,根据表S6中显示的峰面积数据和支持信息中的方程1–3计算了Pt1O6–Ce4+、Pt1O4–Ce4+和Pt1O4–Ce3+–Ov物种的含量。
由于半定量分析,图3c展示了基于各种Pt1On–Ceδ+物种的分布。Pt/CeO2–O600包含60.3%的Pt1O4–Ce4+和39.7%的Pt1O6–Ce4+物种,而Pt/CeO2–O800含有更高比例的65.6% Pt1O4–Ce4+,可能是由于四配位物种的热稳定性较高。值得注意的是,非氧化分散的Pt/CeO2–N600 SACs含有类似的61.4% Pt1O4–Ce4+含量,但富集了约14.2%的Pt1O4–Ce3+–Ov和其余的Pt1O6–Ce4+物种(24.2%)。我们还根据半定量结果计算了Pt/CeO2–N600、Pt/CeO2–O600和Pt/CeO2–O800 SACs的平均Pt电荷状态和CNPt–O,这与EXAFS结果吻合良好(表S7)。这进一步验证了半定量结果的可靠性。基于这些结果,可以推断在氧化/非氧化分散过程中Pt1O4–Ce3+–Ov和Pt1O4–Ce4+之间的转化,并通过DFT计算进一步研究。Ov的重要作用通过促进Pt原子从六配位位点(Pt1O6)迁移到方平面Pt1O4位点(图3d中的(111)晶面或(110)/(100)样的步缘)来揭示。在Ov的帮助下,从Pt1O6到Pt1O4的转化是高度放热的(ΔE = −1.35 eV),而在没有Ov的情况下,这种转化在热力学上是不可行的。
监管Pt 单原子的多样性和反应性
振荡现象在两种不同的Pt/CeO2–N600和Pt/CeO2–O600单原子合金催化剂(非氧化和氧化分散状态)上的CO氧化反应活性中得到展示,如图4a所示。Pt/CeO2–O600表现出较差的低温活性,其轻质点火温度约为200℃。经过随后的600℃ N2处理后,得到的Pt/CeO2–N600表现出显著增强的低温活性,其轻质点火温度为90℃。考虑到Pt单原子物种的分布(图3c),我们发现,在非氧化分散后仅占14.2%的形成的Pt1O4–Ce3+–Ov物种,相比更丰富的Pt1O4–Ce4+和Pt1O6–Ce4+对于低温CO氧化具有更高的位点特异性反应活性。此外,通过循环氧化和非氧化分散,可以可逆地控制这两种不同状态(分布)的Pt/CeO2单原子合金催化剂,导致催化反应活性的振荡(图4a红线与黑线)。

DFT计算被执行以进一步揭示Pt1O4–Ce3+–Ov和Pt1O6–Ce4+作为非氧化和氧化分散的Pt/CeO2 SACs中活性位点的CO氧化反应机理(图4b和表S8)。反应循环始于邻近的氧空位(Pt1O4–Ce3+–Ov),其中CO或O2在Ov上的竞争吸附显示最低的活化能,分别为32.1和63.3 kJ/mol(图4b和S9)。 Pt1O6–Ce4+上的CO氧化遵循类似的反应路径,如图S10所示(蓝线),相应步骤的两个活化能分别为∼38.6和95.6 kJ/mol,与先前报道的分别为34和122 kJ/mol一致(图S10)。因此,DFT计算揭示了邻近的氧空位与附近的活性金属位点协同作用,显著促进了CO吸附/O2活化,并进一步降低了反应壁垒,导致在低温下CO氧化的显著位点特异性反应。
结论
总的来说,通过使用多种先进的表征技术,包括ETEM、STEM和EXAFS,我们在原位识别了Pt/CeO2的氧化和非氧化分散态。尽管非氧化分散的Pt/CeO2–N600和氧化分散的Pt/CeO2–O600 SACs具有类似的Pt电荷状态和CNPt–O,但它们在邻近氧空位上表现出明显的变化,导致产生不同的Pt1–On–Ceδ+单原子物种(即Pt1O4–Ce3+–Ov、Pt1O4–Ce4+和Pt1O6–Ce4+)。通过拉曼光谱和计算模拟,我们已经实现了对给定SACs中不同Pt1On–Ceδ+单原子物种分布的半定量分析。令人惊讶的是,对于低温CO氧化的最高位点特异性反应提供的Pt1O4–Ce3+–Ov物种仅占其他丰富的Pt1O6–Ce4+或Pt1O4–Ce4+物种的总量的14.2%。DFT计算进一步验证了形成的邻近氧空位在非氧化和氧化分散过程中促进六配位(Pt1O6–Ce4+)和四配位位点(Pt1O4–Ceδ+)之间动态转化的机制。因此,我们的结果阐明了给定SAC中各种单原子物种的多样性和分布,并提供了一种有效的方法来调控它们的动态转化和可逆性反应。
实验方法
样品制备
使用文献中描述的方法进行CeO 2粉末的合成,即将Ce(NO3) 3 ·6H2O(Alfa Aesar)在空气中于350℃下煅烧2小时。这铂/CeO2催化剂(1wt%铂,标称)是通过初湿浸渍制备的。具体地,将H2PtCl6·6H2O溶液滴加到CeO2粉末中,然后将混合物在80℃下真空干燥12小时。将所得样品在O2中800℃下煅烧10h,然后在H2气氛下400℃下煅烧4h,得到Pt/CeO2。
氧化和非氧化分散Pt/CeO2
对于氧化分散,Pt/CeO2催化剂在O2气氛中于600和800 ℃下煅烧10 h,分别记为Pt/CeO2–O600 和Pt/CeO2–O800。对于非氧化分散,Pt/CeO2催化剂在N2气氛下于600和800 ℃下煅烧10 h,分别记为Pt/CeO2–N600 和Pt/CeO2–N800。
表征
X-ray absorption spectroscopy (XAS) at Pt LIII-edge including the X-ray absorption near-edge structure (XANES) and extended X-ray absorption find structure (EXAFS) was performed at Beijing Synchrotron Radiation Facility (BSRF) in China. The output beam was selected by a Si(111) monochromator, and the energy was calibrated by a Pt foil. The data were collected at room temperature under fluorescence mode using a Lytle detector. The Athena software package was employed to process the XAS data.
Raman spectroscopy was conducted on a Renishaw (NanoWizard) with 532 nm laser excitation and a power of 0.1 mW.
In situ environmental TEM was carried out in situ on the Titan Themis G3 ETEM (Thermo Scientific Company), which was used at 300 kV with a Cs corrector for parallel imaging (CEOS GmbH) and a measured resolution of better than 1.0 Å. TEM images were recorded by a Gatan OneView camera under an exposure time of 0.5 s. During the in situ dispersion process, the pressure of N2 or O2 was controlled by a mass flow controller and introduced into the microscope that was differentially pumped at a stable octagon pressure (near the specimen) of 1 mbar. The Pt/CeO2 catalysts were loaded onto the chip of a microelectromechanical system (MEMS)-based heating holder (DENS solutions Wildfire) for the in situ heating. The Pt/CeO2 was heated to 100, 600, and 800 °C under 1 mbar N2 with a temperature ramping rate of 10 °C/s. After cooling to 20 °C, the atmosphere was switched to 1 mbar O2 and followed with the same thermal process. The software package Digital Micrograph (GMS 3.22) was employed to analyze the TEM results.
HAADF-STEM imaging was performed on a JEM ARM200F thermal-field emission microscope with a probe Cs corrector working at 200 kV. For the HAADF imaging, a convergence angle of ∼23 mrad and collection angle range of 68–174 mrad were adapted for the incoherent atomic number imaging.
H2 temperature-programmed reduction (H2-TPR) experiments were conducted using an Auto Chem II. First, 50 mg of samples was pretreated in N2 (30 mL/min) at 300 °C for 0.5 h. After cooling the sample to room temperature, the sample temperature was raised from room temperature to 900 °C at a rate of 10 °C/min under a flow of 5%
H2/N2 (30 mL/min). The signals were recorded using a TCD detector.
CO-FTIR was obtained using a Bruker TENSOR27 spectrometer that was equipped with a mercury cadmium telluride (MCT) detector. The sample was heated to 150 °C in N2 (25 mL/min) for 30 min to eliminate surface contamination. Then, the sample was cooled to the room temperature and recorded the background spectrum in Ar. Next, the gas was changed to 10% CO/Ar at the same flow rate for 30 min to reach the CO saturation coverage. Then, the sample was exposed to Ar flow to remove gas-phase CO, and the spectra with a resolution of 4 cm–1 were recorded through 256 scans.
The oxygen storage capacity (OSC) test was conducted using a Shimadzu, GC-14B. First, 100 mg of sample was reduced by 10% H2/Ar at 550 °C for 60 min. Then, the temperature was decreased to 200 °C in pure He for 30 min. Finally, a stream of 5% O2/He (40 mL/min) was injected into the reduced sample until saturation.
X-ray diffraction (XRD) was performed using a DX-2700. The patterns were collected from 20 to 90 °at a speed of 8°/min, operating at 40 kV and 30 mA using a Cu Kα radiation source (λ = 0.15432 nm).
X-ray photoelectron spectroscopy (XPS) was recorded by a Thermo Fisher ESCALAB 250 xi with Al Kα radiation (1486.6 eV). Binding energies were calculated with respect to C 1s at 284.8 eV.
原文地址:Reversible Transformation and Distribution Determination of Diverse Pt Single-Atom Species
声明:仅代表推文作者个人观点,作者水平有限,如有不科学之处,请大家指正!















 1026
1026











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









