







 本文详细介绍了SNP(单核苷酸多态性)检测的几种主要软件工具,包括GATK、FreeBayes、BCFtools、VarScan和DeepVariant。GATK因其高级算法和全面的数据处理流程而被广泛使用,但学习曲线陡峭。FreeBayes使用贝叶斯方法,适用于特定的SNP calling任务。BCFtools专注于处理BCF文件。VarScan适合小规模数据分析。DeepVariant基于深度学习,提供更准确的变异检测。文章还详细阐述了GATK的SNP-calling流程,包括数据预处理、变异检测和校准等步骤,以及如何使用这些工具进行植物基因组SNP和INDEL变异分析。
本文详细介绍了SNP(单核苷酸多态性)检测的几种主要软件工具,包括GATK、FreeBayes、BCFtools、VarScan和DeepVariant。GATK因其高级算法和全面的数据处理流程而被广泛使用,但学习曲线陡峭。FreeBayes使用贝叶斯方法,适用于特定的SNP calling任务。BCFtools专注于处理BCF文件。VarScan适合小规模数据分析。DeepVariant基于深度学习,提供更准确的变异检测。文章还详细阐述了GATK的SNP-calling流程,包括数据预处理、变异检测和校准等步骤,以及如何使用这些工具进行植物基因组SNP和INDEL变异分析。
SNP-calling, genomics, genetics
- calling: GATK, FreeBayes, VarScan,DeepVariant,reads_mapping + samtools mpileup + bcftools
- statistics: BCFtools
1.Software
https://zhuanlan.zhihu.com/p/662413360
1.1 GATK (Genome Analysis Toolkit)
GATK (Genome Analysis Toolkit) 由 Broad Institute 开发,最初为变异检测和基因组分析设计。包含一系列高级算法,用于从DNA测序数据中检测 SNPs、插入/缺失变异 (Indels)、结构变异 (SVs) 等。GATK 提供一系列不同工具和步骤,用于质量控制、变异检测和后续过滤。
优点:
高度可定制:GATK 允许用户根据其研究需求进行高度定制化分析,适用于各种应用。
先进的变异检测:GATK 提供一些高度敏感和精确的变异检测算法,尤其适用于复杂变异的检测。
数据处理流程完整:GATK 提供一整套数据处理工具,包括质量控制、比对、基质检测等。
缺点:
学习曲线陡峭:GATK 需要高的技术知识,初学者较难度
计算资源要求高:一些 GATK 分析需要大量计算资源
更新频繁:GATK 经常发布新版本,需不断更新适应
1.2 FreeBayes
FreeBayes 是一款开源 SNP calling 软件,它专注于变异检测。它使用贝叶斯推断方法来检测 SNP 和 Indels,并具有一些高级功能,如群体 SNP calling和样本混合的处理。
优点:
高度可定制:FreeBayes 具有许多参数选项,允许用户根据需要进行自定义设置。
贝叶斯方法:使用贝叶斯推断方法,可以提供可靠的变异检测结果。
容易上手:相对其他 SNP calling 工具,FreeBayes 较易
缺点:
对计算资源要求较高:仍需要适量计算资源
不适用于所有应用:FreeBayes 更适用于特定类型的数据和问题,不一定适用于所有 SNP calling 任务。
1.3 BCFtools -SNP统计工具
BCFtools 是与 SAMtools 一起开发的一组工具,用于处理 BCF(Binary Call Format)文件,这些文件包含 SNP calling 的结果。BCFtools 提供一系列命令,用于过滤、转换和统计 SNP 数据。
优点:
与 SAMtools 集成:BCFtools 可与 SAMtools 无缝集成,方便数据处理和 SNP calling 的流程。
高效:BCFtools 用于处理大型 SNP 数据集时非常高效。
缺点:
通常需要与其他 SNP calling 工具结合使用:BCFtools 通常用于处理 SNP calling 结果,而不是直接执行 SNP calling。
1.4 VarScan
VarScan 是一款用于检测SNPs和 Indels的工具,特别适用于比对测序数据。它可以用于比对测序和 amplicon测序等不同类型的数据。
优点:
灵活性:VarScan 允许用户根据不同的数据类型和需求进行自定义设置。
高度集成:VarScan 可以与许多常见的测序比对工具集成,使其非常灵活。
缺点:
对数据质量要求较高:VarScan 对数据质量要求较高,需要良好的测序覆盖和质量。
更新较慢:VarScan 的更新速度较慢,不如一些其他工具跟得上最新的研究方法。
选择适合您的 SNP calling 软件工具取决于您的研究需求、技术知识水平和数据类型。每个工具都有其独特的优势和限制,因此在选择时需要综合考虑这些因素。在实际研究中,可能需要尝试不同的工具以确定哪个最适合您的项目。此外,随着时间的推移,这些工具可能会不断发展和改进,因此建议定期查看最新版本和文档。
1.5 DeepVariant
DeepVariant基于谷歌的TensorFlow训练开发,通过卷积神经网络(CNN)模型对已知样本进行训练得到一个可以在低覆盖度测序样本分析时显著提高准确度的软件。该软件18年发表在nbt
文章最后,作者提了点速度方面的问题,DeepVariant的最小硬件环境为16GB RAM,SpeedSeq在72 core/100GB RAM的服务器上单个样品的时间可以降到3小时/样本,但是如果给DeepVariant用上4 GB的显存和CUDA计算,则每个样本的时间可以再降低50%。
最后笔者稍微总结一下,目前看来,基于神经网络的DeepVariant前途一片光明,而speedseq借助于其较快的速度仍有助于其挤占GATK一定的份额。GATK在升级到4.0后算法上有了改进,其黄金标准的地位目前来说依旧不会动摇。未来如何不好说
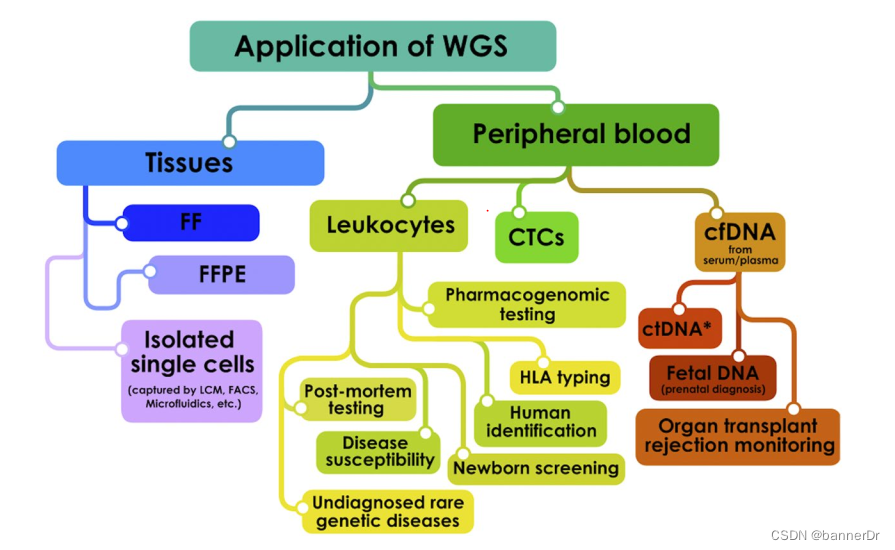
1.6 reads_mapping + samtools mpileup + bcftools
这一套用的是最大似然,比GATK的贝叶斯法快不少
https://www.jianshu.com/p/b4382ca3f1d6
conda install samtools
samtools --version
conda install bcftools
bcftools --version
bwa/bowtie2 - NGS 比对
ALFALFA比对 - 长序列比对(长序列只有IsoSeq?没有其他长序列重测序?)
sam -> bam -> sorted.bam -> bcf -> vcf
conda activate snp
# index
samtools faidx AF04-12_ref.fna
# sam -> bam
samtools view -@ 8 -b -S AF04-12_bgi.sam -o AF04-12_bgi.bam
# bam -> sorted.bam
samtools sort -@ 8 -l 9 -O BAM AF04-12_bgi.bam \
-o AF04-12_bgi.sorted.bam
# sorted.bam -> bcf
samtools mpileup -f ../AF04-12_ref.fna AF04-12_bgi.sorted.bam \
> test.bcf
# bcf -> vcf
bcftools view test.bcf > test.vcf
#https://www.plob.org/article/9824.html
samtools view -bS tmp1.sam > tmp1.bam
samtools sort tmp1.bam tmp1.sorted
samtools index tmp1.sorted.bam
samtools mpileup -d 1000 -gSDf ../../../ref-database/hg19.fa tmp1.sorted.bam |bcftools view -cvNg – >tmp1.vcf
2. ReSeq SNP-calling GATK
https://www.jianshu.com/p/ca1214a0443d
• SNP 强调群体在这个位点有多样性
• Genotype 强调个体在这个位点是哪种基因型。SNV属于genotype的范畴,表示这个genotype 是个突变,独一无二
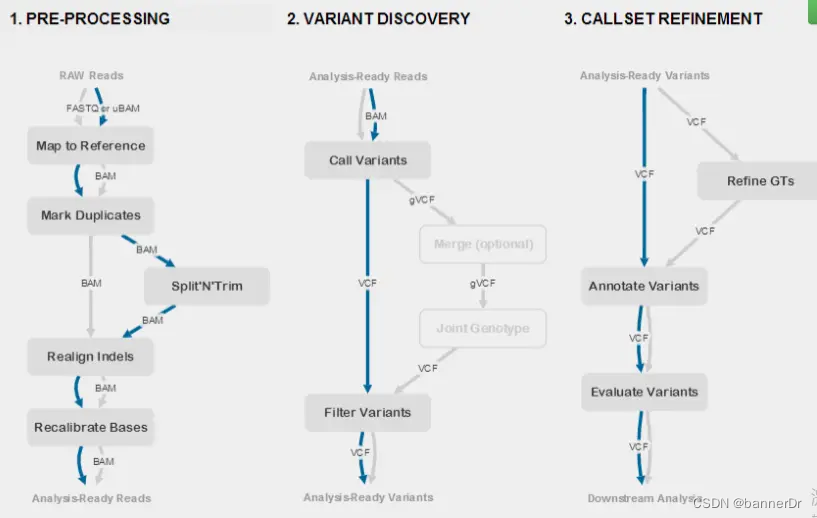
https://gencore.bio.nyu.edu/variant-calling-pipeline-gatk4/
GATK pipeline is intended for calling variants in samples that are clonal – i.e.a single individual. The frequencies of variants in these samples are expected to be 1 (for haploids or homozygous diploids) or 0.5 (for heterozygous diploids). To call variants in samples that are heterogeneous, such as human tumors and mixed microbial populations, in which allele frequencies vary continuously between 0 and 1 researcher should use GATK4 Mutect2 which is designed to identify subclonal events (workflow coming soon).
GATK流程用于call克隆样本中的变异,即单个个体。这些样本中变异的频率预计为1(单倍体或纯合二倍体)或0.5(杂合二倍体)。call异质性样本中的变异,例如人类肿瘤和混合微生物种群,其中等位基因频率在0和1之间连续变化,应使用GATK4Mutect2,该工具识别亚克隆
Base Quality Score Recalibration (BQSR) is an important step for accurate variant detection that aims to minimize the effect of technical variation on base quality scores (measured as Phred scores). As with the original pipeline (link), this pipeline assumes that a ‘gold standard’ set of SNPS and indels are not available for BQSR. In the absence of a gold standard the pipeline performs an initial step detecting variants without performing BQSR, and then uses the identified SNPs as input for BQSR before calling variants again. This process is referred to as bootstrapping and is the procedure recommended by the Broad Institute’s best practices for variant discovery analysis when a gold standard is not available.
碱基质量分数重新校准(BQSR)是准确检测变异的重要步骤,旨在最大限度地减少技术差异对碱基质量分数(以Phred分数测量)的影响。与原始流程一样,该流程假定BQSR没有“金标准”的snp/indel。在没有金标准的情况下,流程在不执行BQSR的情况下执行检测变异的初始步骤,然后在再次calling variants之前使用已识别的snp作为BQSR的输入。这个过程被称为bootstrapping,并且是Broad研究所在没有黄金标准时推荐的用于variant 发现分析的最佳实践的过程。
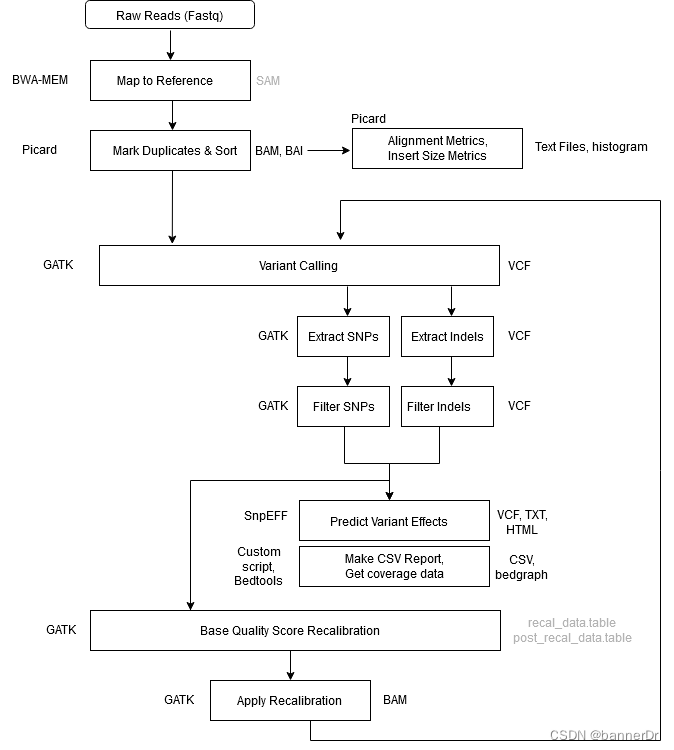
2.1 流程总结
• bwa,samtools,picard。ref.fa建立索引,bwa将fastq比对回参考基因组,生成sam(小于10M,使用bwa的-is参数,参考基因组很大,使用bwtsw参数)
• 使用picard对sam文件排序后生成bam文件(使用samtools会删除重复位点,而picard只会标记重复并不删除)。使用samtools对bam建立索引。利用已经注释过的indel.vcf文件对indel周围realignment,这个操作有两个步骤,第一步生成需要进行realignment的位置信息,第二步 才是对这些位置realignment,最后生成一个重排好的BAM。
• 接下来对mapping得到的bam文件进行矫正,消除偏态,去除假阳性,这里主要针对测序质量不是特别好的数据,可以根据fastqc结果对测序结果进行解读,如果测序质量比较好的话可跳过。
• 使用HaplotypeCaller发现变异并生成gvcf文件。随后使用CombineGVCFs将不同文件整合,GenotypeGVCFs函数输出vcf文件。
• 下一步使用VariantRecalibrator校准,并根据可视化结果确定最佳参数,确定参数之后再运行一次VariantRecalibrator得出结果
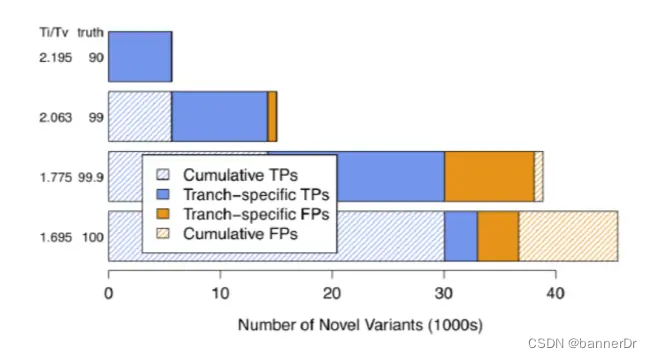
• 如何确定参数:(1) an注释信息选择:从图中可以看出,这个模拟结果可以很好的将真实的变异位点和假阳性变异位点分开,形成明显的界限, 也就是说,如果一个变异位点的这两个注释值,只要有一个落在了界限之外,就会被过滤掉。最主要的是要 看右边两个图片,只要能很好的区分开novel和known以及filtered和retained就可以。其实在如何选择注释值 存在一定得主观性,因此,在做VariantRecalibrator时可以做两次,第一次尽可能的多的选择这些注释值, 第一遍跑完之后,选择几个区分好的,再做一次VariantRecalibrator,然后再做ApplyRecalibration。 (2) tranche值的设定:如果这个值设定的比较高的话,那么最后留下来的变异位点就会多,但同时假阳性的位点也会相应增加; run两遍VariantRecalibrator,第一遍的时候多写几个阈值,看哪个阈值结果好,选择一个最好的阈值,再run一遍 VariantRecalibrator。 区分标准: 1. 看结果中已知变异位点与新发现变异位点之间的比例,这个比例不要太大,大多数新发现的变异都是假阳性,如果太多的 话,可能假阳性的比例就比较大; 2. 看保留的变异数目,这个就要根据具体的需求进行选择了。 – 3. 看TI/TV值,对于人类全基因组,这个值应该在2.15左右,对于外显子组,这个值应该在3.2左右,不要太小或太大,越接近 这个数值越好,这个值如果太小,说明可能存在比较多的假阳性。 千人中所选择的tranche值是99,仅供参考。
• 最后使用annovar对vcf进行注释并可视化结果
2.2.1 索引
# 基因组索引
#首先建立参考基因组索引
参数-a用于指定建立索引的算法:
bwtsw 适用于>10M
is 适用于参考序列<2G (默认-a is)
可以不指定-a参数,bwa index会根据基因组大小来自动选择合适的索引方法
##bwa
bwa index -a is ./ref/ 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 526
526

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









