






fastq-dump是大家经常使用的,主要是由于从NCBI中下载的数据格式大部分是SRA格式,此时就需要使用fastq-dump将SRA格式转换成为fasta格式
fastq-dump是sratoolkit软件中的一个功能,首先安装sratoolkit
打开ncbi官网,点击Download--Download Tools--


点击Download,选择对应的版本
获取sratoolkit的下载地址
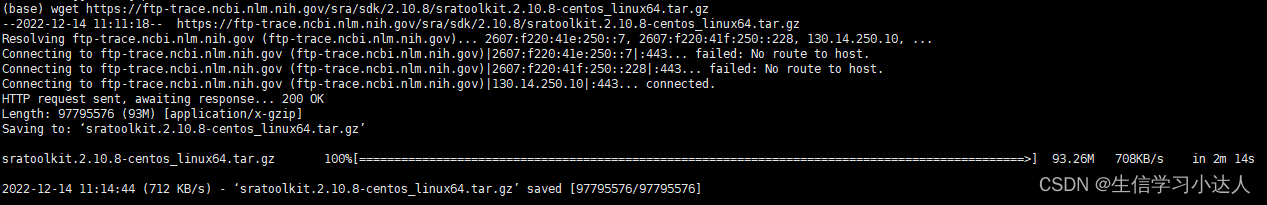
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.10.8/sratoolkit.2.10.8-centos_linux64.tar.gz
sratoolkit安装和使用
tar zxvf sratoolkit.2.10.8-centos_linux64.tar.gz
cd sratoolkit.2.10.8-centos_linux64/bin
vdb-config -i #配置存储位置
配置环境变量,方便其在任何位置均可以使用
vi ~/.bashrc #用vi/vim编辑器修改bashrc文件
i #由command line进入insertion line
export PATH=$PATH:/home/urname/local/app/sratoolkit/bin
ESC, :wq #退出vi编辑器并保存文件
source ~/.bashrc #让配置生效
使用方法也相简单
prefetch SRR1036346 # SRR1036346为你想要获取的sra数据编号
# fastq_dump可将sra数据转化为fastq格式数据
fastq_dump --split-e SRR1036346.sra
# fasterq_dump支持多线程
fasterq_dump --split-3 SRR1036346.sra -e 10 -o SRR1036346
# 若为双端数据,则会产生两个数据,分别为SRR1036346_1.fastq 和 SRR1036346_2.fastq;若为单端数据,则只有一个数据,为SRR1036346.fastq.
我们一般使用fastq-dump的方式为
fastq-dump --split-3 SRR103346双端测序结果将保存到一个文件里, 但是如果你加上--split-3之后, 他会把原来双端拆分成两个文件,但是原来单端并不会保存成两个文件. 另外,你用--gzip就能输出gz格式, 同时进行压缩。

















 2527
2527

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









