







IBD分析(IdentitybyDescentAnalysis)是一种遗传学分析方法,用于研究个体间或群体中共享的祖先片段。IBD片段是指两个或多个个体从共同祖先继承的、未经过重组的相同DNA片段。通过识别这些片段,可以推断个体间的亲缘关系、群体结构以及进化历史。地理隔离(IBD)和适应性隔离(IBE)是造成分化的两种主要原因
1.IBD分析的核心概念:
1.IBD片段:两个个体从共同祖先继承的相同DNA片段。片段长度越长,表明共同祖先越近;片段长度越短,表明共同祖先越远。
2.IBS(IdentitybyState):个体间在某些位点上具有相同的等位基因,但这些基因不一定来自共同祖先。IBD是IBS的一个子集,IBD片段一定是IBS,但IBS片段不一定是IBD。
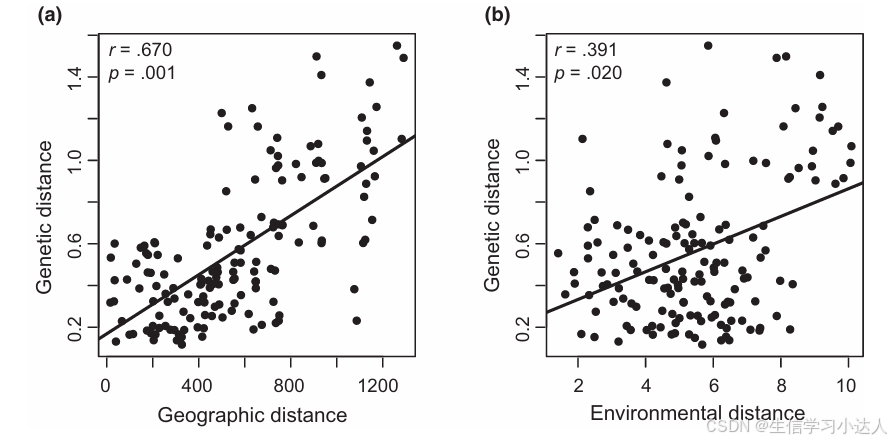
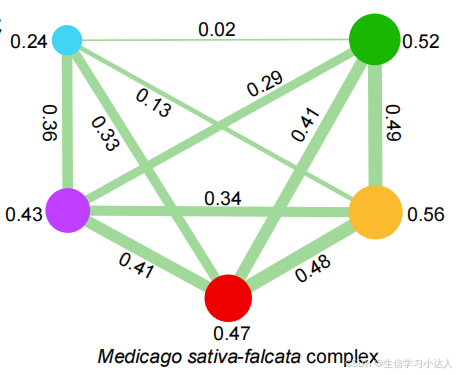
图片来源于(DOI:10.1016/j.molp.2024.04.013)
2.IBD分析的应用:
1.亲缘关系推断:通过检测IBD片段,可以推断个体间的亲缘关系(如兄弟姐妹、堂表亲等)。
在家族遗传学中用于构建家系图。
2.群体遗传学研究:分析群体中IBD片段的分布,推断群体的历史(如瓶颈效应、迁移事件等)。研究群体间的遗传分化程度。
3.复杂疾病研究:通过检测患病个体间共享的IBD片段,定位与疾病相关的基因区域。在全基因组关联分析(GWAS)中用于提高统计效力。
4.育种中的应用:在动植物育种中,利用IBD分析筛选优良亲本,优化育种策略。通过检测IBD片段,评估个体的遗传多样性。
3.IBD分析的步骤:
1.数据准备:获取高质量的基因型数据(如SNP芯片数据或全基因组测序数据)。对数据进行质量控制(如去除低质量位点、填补缺失数据)。
2.IBD片段检测:使用算法(如PLINK、GERMLINE、BEAGLE等)检测个体间共享的IBD片段。根据位点的等位基因频率和连锁不平衡(LD)信息,区分IBD和IBS。
3.结果分析:计算个体间共享的IBD片段长度和数量。构建亲缘关系矩阵,推断个体间的遗传关系。可视化IBD片段的分布(如染色体图谱)。
4.统计推断:利用IBD片段信息进行群体结构分析、历史人口统计推断或疾病关联分析。
利用plink软件进行IBD计算
##step 1, filter SNP by LD
perl /public/agis/zhouyongfeng_group/zhangfan02/vcf_file/lecture06_07_add_id.pl merge_712_allele2_meanDP3-62_miss0.2.vcf.recode.vcf merge_712_allele2_meanDP3-62_miss0.2_addid.vcf.recode.vcf
plink --vcf merge_712_allele2_meanDP3-62_miss0.2_addid.vcf.recode.vcf --indep-pairwise 100 50 0.2 --out m.714.snp.ld --allow-extra-chr --make-bed
plink --bfile m.712.snp.ld --extract m.712.snp.ld.prune.in --out all_712_iqtree_plink_vcf --recode vcf --allow-extra-chr
plink --vcf all_712_iqtree_plink_vcf.vcf --keep 702_accessions.txt --allow-extra-chr --threads 1 --recode vcf --out all_702_iqtree_plink_vcf
##step 2, PCA analysis
plink --allow-extra-chr --threads 2 --vcf all_702_iqtree_plink_vcf.vcf --pca 20 --out 702_accession_pca
#please refer PCA.R for plot
##step 3, IBD analysis
plink --vcf all_702_iqtree_plink_vcf.vcf --const-fid --allow-extra-chr --genome --out IBD_702_result --threads 2
#please refer IBD.R for plot
IBD<-read.table("IBD_702_result.genome",header=TRUE,sep="")
group_info=read.table("group.txt",header=TRUE,sep="")
colnames(group_info)=c("IID1","group")
library(dplyr)
combine1=inner_join(IBD, group_info, by="IID1")
colnames(group_info)=c("IID2","group2")
combine2=inner_join(combine1, group_info, by="IID2")
result=aggregate(x=combine2$PI_HAT,by=list(combine2$group,combine2$group2),FUN=mean)
colnames(result)=c("group1","group2","mean_PI_HAT")
write.csv(result,"IBD_result.csv",row.names = F) ##used for cytoscape
##I use cytoscape for plot, it is a window interface software. please refer:https://cytoscape.org/manual/Cytoscape3_7_0Manual.pdf.备注:重测序数据的下载、质控、比对排序去重复、Calling-SNPs(GATK软件)最终获得merge_712_allele2_meanDP3-62_miss0.2_addid.vcf.recode.vcf这个SNP的vcf文件
4.常用工具和软件:
1.PLINK:常用的遗传数据分析工具,支持IBD片段检测和亲缘关系分析。
2.GERMLINE:高效的IBD片段检测工具,适用于大规模数据集。
3.BEAGLE:基于隐马尔可夫模型(HMM)的IBD检测工具,适用于高密度基因型数据。
4.KING:用于亲缘关系推断和IBD分析的工具。
总结:
IBD分析是一种强大的遗传学工具,广泛应用于亲缘关系推断、群体遗传学、疾病研究和育种等领域。通过检测个体间共享的祖先片段,IBD分析能够揭示遗传背景、进化历史和复杂性状的遗传基础,为遗传学和育种研究提供重要支持。
参考来源:

















 227
227

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









