






用blastall进行序列比对
blastall是最常用的blast程序之一,一般使用的参数如:-p、-i、-d、-o、-e等几个。
-p: 执行的程序名称
-d: 搜索的数据库名称
-i : 要查询的序列文件名(Query File)
-e:(数学)期望值(Expectation value),E值是个统计阈值,缺省值10, 意指比对结果中由于随机偶然性产生的匹配结果不大于10,E值越小结果越可靠。
-o :查询结果输出文件名
-m: 比对结果显示格式选项,缺省值为0 ,即pairwise格式。另外还可以根据不同的需要选择1~6等不同的格式。
-I :在描述行中显示gi号[T/F],缺省值F
-v :单行描述(one-line description)的最大数目,缺省值500
-b :显示的比对结果的最大数目,缺省值250
-F :对于要查询的序列做低复杂度区域(low complexity regions, LCR)的过滤[T/F],缺省值T。对blastn用的是DUST程序,其他比对用的是SEG程序。
所谓“低复杂度区域”是指某些或一些残基过多表现,短周期重复等。对于高等哺乳动物的基因组序列,可以先用RepeatMask程序遮蔽重复元件。在输出结果中,对LCR区的序列核酸用“N”代替,蛋白质序列用“X”代替。
-a:运行BLAST程序所使用的处理器的数目,缺省值1
-S:在数据库中搜索时所使用的核酸链(strand),只对blastn、blastx和tblastx有效;1表示top,2表示bottom,3表示both;缺省值3
-T: 产生HTML格式的输出[T/F],缺省值F
-n: 使用MegaBlast搜索[T/F],缺省值F
-G: 打开一个gap的罚分(0表示使用缺省设置值),默认0
-E: 扩展一个gap的罚分(0表示使用缺省设置值),默认0
-q: 一个核酸碱基的错配(mismatch)的罚分(只对blastn有效),缺省值-3
-r : 一个核酸碱基的正确匹配(match)的奖分(只对blastn有效),缺省值1
-M: 所使用的打分矩阵,缺省值BLOSUM62
1.参数说明
基本参数、比对优化参数、结果输出参数、控制输入参数
| 参数 | 说明 | 值 | 默认值 | 备注 |
| -p | 使用的程序 | 字符[String] | blastn blastp blastx tblastn tblastx | |
| -d | 使用的数据库 | 文件名[File In] | nr | |
| -i | 搜索用的序列 | 文件名[File In] | stdin | |
| -e | 期望值 | 数字[Real] | 10.0 | |
| -m | 控制比对结果的样式 | 0到11的整数[Integer] | 0 | 0 = pairwise, 1 = query-anchored showing identities, 2 = query-anchored no identities, 4 = flat query-anchored, no identities, 5 = query-anchored no identities and blunt ends, 6 = flat query-anchored, no identities and blunt ends, 7 = XML Blast output, 8 = tabular, 9 tabular with comment lines 10 ASN, text 11 ASN, binary |
| -o | 比对结果存放的文件名 | 文件名[File Out] | stdout | |
| -F | 过滤询问序列 | [String] | T | DUST with blastn, SEG with others |
| -G | 打开gap得分 | [Integer] | -1 | |
| -E | 延伸gap得分 | [Integer] | -1 | |
| -X | X dropoff value for gapped alignment (in bits) | [Integer] | 0 | blastn 30, megablast 20, tblastx 0, all others 15 |
| -I | 显示gi号Show GI’s in deflines | [T/F] | F | |
| -q | 核酸错配罚分 | [Integer] | -3 | blastn only |
| -r | 核酸匹配得分 | [Integer] | 1 | blastn only |
| -v | Number of database sequences to show one-line descriptions for (V) | [Integer] | 500 | |
| -b | Number of database sequence to show alignments for (B) | [Integer] | 250 | |
| -f | Threshold for extending hits | [Integer] | 0 | blastp 11, blastn 0, blastx 12, tblastn 13, tblastx 13, megablast 0 |
| -g | Perform gapped alignment | [T/F] | T | not available with tblastx |
| -Q | 指定询问序列使用的遗传密码 | [Integer] | 1 | |
| -D | 指定数据使用的遗传密码 | [Integer] | 1 | for tblast[nx] only |
| -a | 使用CPU的数目 | [Integer] | 1 | |
| -O | SeqAlign file | [File Out] | 可选 | |
| -J | Believe the query defline | [T/F] | F | |
| -M | 比对使用的矩阵 | [String] | BLOSUM62 | |
| -W | Word size | [Integer] | 0 | blastn 11, megablast 28, all others 3 |
| -z | 数据库的有效长度Effective length of the databas | [Real] | 0 | use zero for the real size |
| -K | Number of best hits from a region to keep | [Integer] | 0 | off by default, if used a value of 100 is recommended |
| -P | 0 for multiple hit, 1 for single hit | [Integer] | 0 | does not apply to blastn |
| -Y | Effective length of the search space | [Real] | 0 | use zero for the real size |
| -S | Query strands to search against database | [Integer] | 3 | for blast[nx], and tblastx, 3 is both, 1 is top, 2 is bottom |
| -T | 将结果保存为HTML格式 | [T/F] | F | |
| -l | 通过gi号列表,限制搜索范围 | [String] | Optional | |
| -U | Use lower case filtering of FASTA sequence | [T/F] | Optional | |
| -y | X dropoff value for ungapped extensions in bits | [Real] | 0.0 | 0.0 invokes default behavior blastn 20, megablast 10, all others 7 |
| -Z | X dropoff value for final gapped alignment in bits | [Integer] | 0 | blastn/megablast 50, tblastx 0, all others 25 |
| -R | PSI-TBLASTN checkpoint file | [File In] | Optional | |
| -n | MegaBlast search | [T/F] | F | |
| -L | Location on query sequenc | [String] | Optional | |
| -A | Multiple Hits window size | [Integer] | 0 | default if zero (blastn/megablast 0, all others 40) |
| -w | Frame shift penalty | [Integer] | 0 | OOF algorithm for blastx |
| -t | Length of the largest intron allowed in a translated nucleotide sequence when linking multiple distinct alignments | [Integer] | 0 | 0 invokes default behavior; a negative value disables linking. |
| -B | Number of concatenated queries | [Integer] | 0 | for blastn and tblastn |
| -V | Force use of the legacy BLAST en gine | [T/F] | F | Optional |
| -C | Use composition-based statistics for tblastn | [String] | D | D or d: default (equivalent to F) 0 or F or f: no composition-based statistics 1 or T or t: Composition-based statistics as in NAR 29:2994-3005, 2001 2005, conditioned on sequence properties 3: Composition-based score adjustment as in Bioinformatics 21:902-911, 2005, unconditionally For programs other than tblastn, must either be absent or be D, F or 0. |
| -s | Compute locally optimal Smith-Waterman alignments | [T/F] | F | This option is only available for gapped tblastn. |
2. 使用说明与示例
程序使用说明

3.blast格式
经常使用blast,一般使用-m 8格式作为blast结果的,但是blast的-m 8结果竟然没有标题,那就记录一下
首先展示-m 8结果文件如下:
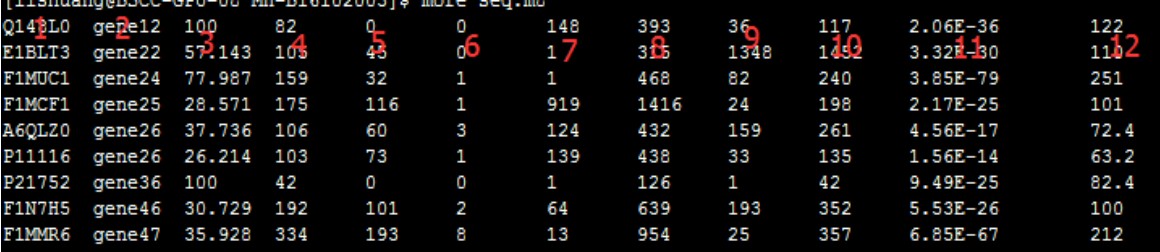
从图中可以看出共12列,下面来列举一下这12列的意思
1、Query id:查询序列ID标识
2、Subject id:比对上的目标序列ID标识
3、% identity:序列比对的一致性百分比
4、alignment length:符合比对的比对区域的长度
5、mismatches:比对区域的错配数
6、gap openings:比对区域的gap数目
7、q. start:比对区域在查询序列(Query id)上的起始位点
8、q. end:比对区域在查询序列(Query id)上的终止位点
9、s. start:比对区域在目标序列(Subject id)上的起始位点
10、s. end:比对区域在目标序列(Subject id)上的终止位点
11、e-value:比对结果的期望值,解释是大概多少次随即比对才能出现一次这个score,Evalue越小,表明这种情况从概率上越不可能发生,那么发生了即说明这更有可能是真实的相似序列
12、bit score:比对结果的bit score值
一般情况我们看第3、11、12两列,e值越小越可靠。
blast对应的参数是 -m 8
blast+对应的参数是-outfmt 6

















 2947
2947

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









