






 文章详细阐述了转录组测序后如何分析基因全长,包括使用BLAST工具进行核苷酸和氨基酸序列比对,确定基因结构,预测信号肽和保守结构域,进行多序列比对以及构建进化树。此外,还介绍了原核表达中的引物设计和同源重组过程。
文章详细阐述了转录组测序后如何分析基因全长,包括使用BLAST工具进行核苷酸和氨基酸序列比对,确定基因结构,预测信号肽和保守结构域,进行多序列比对以及构建进化树。此外,还介绍了原核表达中的引物设计和同源重组过程。
转录组测序完成之后,转录组分析筛选出某些基因。
针对这些基因的分析……
首先,这条序列是不是全长?获得了全长的序列才有预测的意义。
复制关于此序列的所有核苷酸碱基,
到ncbi——BLAST——blastx (输入核苷酸序列查询氨基酸序列)
最上方的一条序列即与之最相近。
打开最相近的序列,

这显示你所查询的序列第1位与目标序列的第56位对应上了,说明5'端(N端)缺少50个左右核苷酸,此序列不是全长序列。
通常来讲,转录组测序出来的序列全长可用于分析,不过我们一般进行验证全长后进行分析。
若没有验证全长的情况下,转录组测序得到的全长也可用于序列分析(相差无几)。
复制整段序列,在 ExPASy - Translate tool 网页内翻译氨基酸,把M、- - 上方及M、- -前一个氨基酸上方的6个左右核苷酸复制,在全长序列中查找,标出起始子和终止子。
eg:CSP
复制CSP全长序列,粘贴至下列网址

- - 代表终止子
把- - 上方及- -前一个氨基酸上方的6个左右核苷酸复制,在全长序列中查找,标出终止子。
(1)基因结构——外显子/内含子
方法一、ncbi——blast——nucletide blast
复制粘贴包括起始子及终止子前后的UTR全部核苷酸序列,数据库选择wgs

物种选择 Locusta migratoria (taxid:7004)
blast 查询序列与基因组片段位置一一对应比较。
点击图谱选择最全的一条序列,将其他取消选择,然后点第三栏Alignment
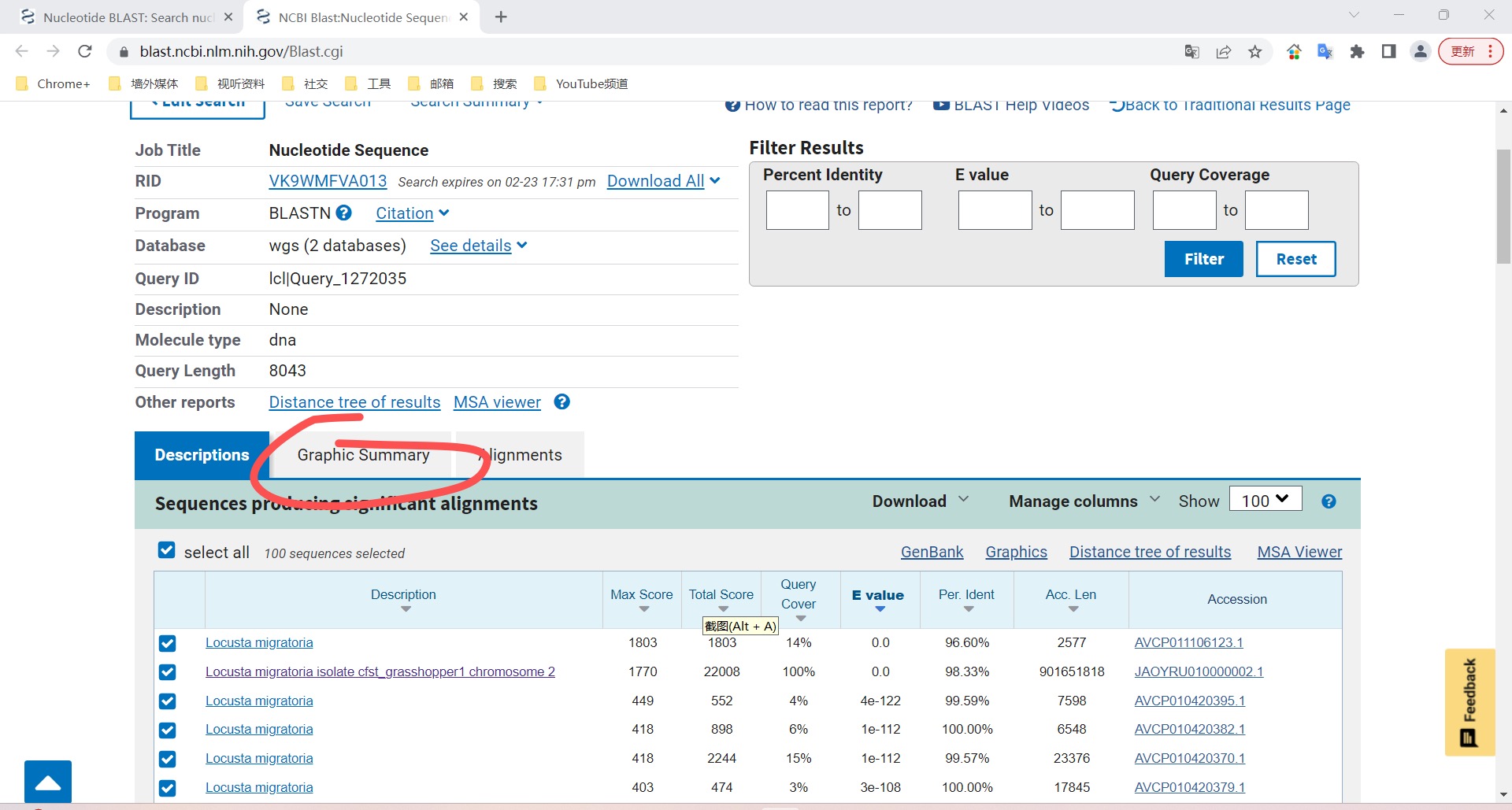
点击图谱选择最全的一条序列,将其他取消选择,然后点第三栏Alignment
Sort by Query start position
记录Query1 和Query最后1个对应的subject基因组序列位置(红色),看正向还是反向
右下角的**load next set**一定要点!!!否则序列不全!!!
选择ATG上游3000 TGA下游1000 bp左右 region下载
与自己基因序列**全长**(ATG - TGA)
方法二、飞蝗数据库 网址 LocustBase-Locust Genome data(康乐老师)
LocustBase——BLAST
选择locust genome数据库,basic search
将比对最好的基因组序列下载下来,包括FASTA格式以及带数字标记的序列(复制下来)。
打开Snapgene,把基因组序列粘贴,OK


再新建一个file,粘贴你的全长序列,保存这个.dna文件到桌面
接着打开全基因组的那个文件,在工具中,

导入刚刚那个.dna文件,选中比对上的序列(只有基因组序列可选),然后,

标记特征 eg: Exon1,可标记颜色等。

选中此段外显子即可获得外显子长度及位置信息,可开始AI 作基因结构图。
(2)核苷酸——氨基酸序列分析
复制 从起始密码子ATG到终止子
 在ExPASY translate tool 中
在ExPASY translate tool 中

此选项出来只有氨基酸序列
ExPASY translate tool ——Includes nucleotide sequences, no spaces——TRANSLATE!
复制5'-3' Frame1 粘贴到新建word(字体预设:Courier New)
开始——清除所有格式
全选——字体Courier New 字号7
手动对齐 一行60字符 (序列长,一行120字符;序列短,一行60字符),若是验证全长后的序列,前后加上转录组5',3'-UTR,未经验证的全长序列可不加5',3'-UTR,每行后标记加和字符数。
全选——右键——字体——全部大写字母
标记CSP序列保守区(4个典型Cys保守区)颜色 字体白色
起始,终止密码子字体红色
段落设置——取消如果定义了文档网格,则与网格对齐
加标注与标题(基因名称)
截图到PPT
信号肽预测 蛋白质信号肽预测 - 在线工具 - 纽普生物 - NovoPro
蛋白质理化特性 复制粘贴aa序列 ProtParam 获得理化特性相关信息
画氨基酸domain,aa domain预测


预测信号肽位置及保守结构域的aa
根据上述,AI作图,氨基酸结构。
NCBI Conserved Domain Search https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
多序列比对(后续2023 用Gendoc进行多序列比对更简单)
从起始密码子到终止密码子复制核苷酸序列,到ncbi——blast——blastx,查询匹配度高的氨基酸序列。
FASTA 序列号
下载一些相似度较高的典型物种,比如同目不同种,不同目的aa序列。
比如:
直翅目:飞蝗、沙漠蝗虫、亚洲小车蝗、黄胫小车蝗、黄脊竹蝗
鞘翅目:赤拟谷盗
鳞翅目:家蚕
膜翅目:松毛虫赤眼蜂
双翅目:番石榴果实蝇、果蝇、冈比亚按蚊、埃及伊蚊
半翅目:稻绿椿
同翅目:扶桑绵粉蚧
先每个目多选几条序列大致比对,然后从中挑选序列,重新比对。

Multiple Sequence Alignment - CLUSTALW
Multiple Sequence Alignment - CLUSTALW https://www.genome.jp/tools-bin/clustalw
复制3条以上aa序列,在clustalw中进行execute multiple alignment
下载 clustalw.aln 文件。
Expasy——swiss model
粘贴aa,search for template——build models 下载.pdb文件
网页——espript3.0
RUN ESPRIPT——Aligned file 选择文件.aln,下一个选择文件.pdb,Number of columns改成85(每行显示85个aa),color scheme选择B&W黑色,output format选 postscript file,PDFfile,TIFF image(600 dpi),可指定顺序,Gap between blocks 3~5(4)——SUBMIT——下载TIFF文件。
多序列比对完成。AI修改。

2023 多序列比对
飞蝗和其他物种的序列保守性分析 aa序列相似度分析
Blastx organism 限定物种
飞蝗 赤拟谷盗 家蚕 冈比亚按蚊 果蝇 小菜蛾 灰飞虱 蚤状蚤 杨叶甲
棉蚜 桃蚜 玉米螟 草贪 蓟马
蜻蜓 蜜蜂 螳螂 七星瓢虫 赤眼小蜂
网柄菌 斑马鱼 人

GeneDoc - File - import .txt -done,





多重序列比对图片导出,点击“Edit”-“Select Blocks for Copy”。
然后再点击“Edit” -“Copy Select Blocks to”-“MetaFile”,即可直接复制得到多重序列比对的图片。
建树 MEGA软件使用
复制自己序列全长 ATG-TGA
ncbi——BLAST——blastx (输入核苷酸序列查询氨基酸序列),fasta格式(>),直接BLAST,查询匹配度高的氨基酸序列。
玉米螟Ostrinia furnacalis、草贪Spodoptera frugiperda、棉铃虫Helicoverpa armigera、斜纹夜蛾Spodoptera litura、家蚕Bombyx mori、印度谷斑螟(Plodia interpunctella)、黑脉金斑蝶Danaus plexippus、棉红铃虫Pectinophora gossypiella、菜青虫Pieris rapae
、大蜡螟Galleria mellonella、甜菜夜蛾Spodoptera exigua、小菜蛾Plutella xylostella
再限定物种 找找其他物种如飞蝗、桃蚜、棉蚜。

下载Fasta格式 .txt
全部序列 放在一个txt中;另存为一份,更改物种-蛋白命名。
MEGA7.0
1.打开软件
2.点工具栏中Align,点下拉菜单中“Edit/build alignment”,选择“Great a new alignment”——OK
在下一对话框中选择“protein”
复制更改命名的fasta格式的全部序列 eg 15条,ctrl+V.

比对后保存,Data——Export Alignment——MEGA Format.(.meg格式)
.meg格式文件拖入MEGA软件框内,构树——Neighbor-Joining Tree

Test of phylogeny选择步长检验Bootstrap method,No. of Bootstrap Replications选择1000;
Model/Method选择p-distance;
Gaps/Missing Data Treatment 选择 Partial deletion, 50。
Bootstrap concensus tree是经过1000自展值综合考量后的发育树。
保存 .mtsx

更改树的形状。
3.打开.mtsx格式的文件,复制聚类树,粘贴在PPT中。
在PPT中全选,取消组合,更改斜体。
图片另存为.png。
找启动子
打开JASPAR
选择insecta数据库,输入CncC,search;
 选中搜到的序列,点开scan,输入下游基因的fasta格式(>),Relative profile score threshold默认80%,scan;
选中搜到的序列,点开scan,输入下游基因的fasta格式(>),Relative profile score threshold默认80%,scan;
+链直接复制标记在上2000下500 word中标记,-链反向互补后在上2000下500标记;
这种结合位点分析方法灵敏度很高,在大多数情况下能检测到有功能的结合位点。但是,大多预测的位点虽在体外(in vitro)能与TF结合,在体内(in vivo)可能并无功能,预测结果仍需做进一步的验证。
对于研究较多的明星基因,ncbi搜基因名,点开基因图谱——tools——sequences text view——annotated选none,粉色代表外显子,绿色代表内含子,紫色代表5',3'-UTR区,白色代表两个基因之间的间隔。
promoter区扩增:选取5'-UTR附近的区域至ATG下游200bp左右进行引物设计
输入全长序列,search

在设计的引物前加上酶切位点和保护碱基
基因组(邢嘉乐d4高)为模板进行普通PCR——UGT392A1 promotor全长。
胶回收连接转化送测序,提质粒UGT392A1 promotor-T3-transT1。
双酶切,连pGL3.0载体,①UGT392A1 promotor-pGL3.0-transT1。无内毒素提质粒。
②CncC-pIEX4-transT1过表达体系,无内毒素提质粒。
①②共转染细胞。
原核表达:
1)网页设计引物
2)snapgene检查是否编码正确氨基酸:
新建 ATG-去除信号肽-去掉终止子序列,前后加上6位酶切位点,保存为9A;
打开载体序列,

选择载体酶切位点(大片段),插入片段选择加酶切位点的序列及酶切位点,与 载体酶切位点顺序一致,clone,保存为pET28a-9A。

从前面的酶切位点开始检查,氨基酸翻译是否正确。
原核表达同源重组引物
https://crm.vazyme.com/cetool/singlefragment.html

载体序列方向不在意,只要5' 3' 接壤的酶切位点选择正确,就没问题。
Snapgene中,EcoR1和Hind3这种酶切位点碱基整体保留,只delete中间的碱基,然后插入自己的片段,看翻译是否正确。

lncRNA必知必会的数据库资源大全 - 曾健明的文章 - 知乎 https://zhuanlan.zhihu.com/p/357635131

















 5686
5686

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









