






 本文开发了新型深度学习框架TRAmHap,结合卷积与递归神经网络,利用DNA甲基化特征预测基因表达。研究筛选平均甲基化、MHL和MCR为模型输入,经实验验证,TRAmHap在预测准确性上优于现有方法,还可预测不同组织和疾病条件下的基因表达。
本文开发了新型深度学习框架TRAmHap,结合卷积与递归神经网络,利用DNA甲基化特征预测基因表达。研究筛选平均甲基化、MHL和MCR为模型输入,经实验验证,TRAmHap在预测准确性上优于现有方法,还可预测不同组织和疾病条件下的基因表达。
期刊:Briefings in Bioinformatics
代码链接:https://github.com/SQ-Gao/TRAmHap
CpG是胞嘧啶(C,Cytosine),磷酸(p,phosphoric acid),鸟嘌呤(G,Guanine )的缩写,也可以去掉磷酸直接叫CG。在哺乳动物中,在基因组中富含GC和CpG的序列区段,叫CpG岛(CpG islands)。
Pearson’s correlation coefficient(皮尔森相关系数),是一种线性相关系数,是最常用的一种相关系数。记为r,用来反映两个变量X和Y的线性相关程度,r值介于-1到1之间,绝对值越大表明相关性越强。
leave-one-out strategy(留一法交叉验证)是一种用来训练和测试分类器的方法,会用到图像数据集里所有的数据,假定数据集有N个样本(N1、N2、Nn),将这个样本分为两份,第一份N-1个样本用来训练分类器,另一份1个样本用来测试,如此从N1到Nn迭代N次,所有的样本里所有对象都经历了测试和训练。
使用这种方法,我们将数据集随机分成10份,使用其中9份进行训练而将另外1份用作测试。该过程可以重复10次,每次使用的测试数据不同。
一、介绍
脱氧核糖核酸(DNA)甲基化(DNAm)是一种重要的表观遗传机制,在染色质结构和转录调控中发挥作用。研究 DNAm 与基因表达之间的关系对于理解其在转录调控中的作用具有重要意义。文中的新型深度学习框架 TRAmHap就是为了研究DNAm与基因表达之间的相关性。
脱氧核糖核酸(DNA)甲基化(DNAm)是一种基本的表观遗传修饰,通过转座元件抑制、基因印记和 X 染色体失活等机制,在胚胎发生和衰老等多种生物过程中发挥着关键作用。DNAm 指的是在胞嘧啶环的碳 5 位置上添加一个甲基,从而形成 5-甲基胞嘧啶,通常发生在基因组中 60-90% 的 CpG 位点上,但位于启动子区域的 CpG 岛(CGI)除外,这些位点在正常细胞中通常是未甲基化的。在癌症中,启动子区 CGI 的高甲基化可导致抑癌基因被抑制,促进肿瘤发生。
在本研究中,开发了一种新型深度学习框架 TRAmHap,它结合了卷积神经网络和递归神经网络,利用启动子近端(TSS 周围 ±2.5 kb)和含增强子的远端区域(TSS 周围 ±25 kb)的平均甲基化和 mHap 水平汇总统计的特征预测基因表达。我们的模型在预测准确性方面明显优于现有方法,并且能够预测不同组织和疾病条件下的基因表达。我们的研究结果表明,基因表达不仅受TSS附近区域(±2.5 kb)DNAm的影响,还受远端增强子的影响,尤其是在基因内染色质相互作用的情况下。
二、材料和方法
1、数据处理
我们利用各种公共数据集进行研究。正常组织数据集来自 ENCODE 项目联盟,包括 15 个人类组织样本、7 个小鼠前脑样本、7 个小鼠心脏样本和 5 个小鼠肝脏样本。我们对这些样本进行了 RNA-seq 和 WGBS 分析。此外,我们还从 NCBI GEO 获得了食管鳞状细胞癌(n = 10)和正常样本(n = 9)的 WGBS 和 RNA-seq 数据,登录号为 GSE149612 (补充表 S1)。每个组织的增强子数据从 EnhancerAtlas 2.0 和 FANTOM5 中下载。超级增强子从 SEdb 2.0 和 SEA 3.0 中下载。预处理后的 ChIA-PET 数据从 GEO 下载,登录号为 GSE90557 。
亚硫酸酯测序数据的适配器使用默认参数的Trim Galore进行裁剪。使用BSMAP将修剪后的reads定位到人类基因组版本hg19,参数为:' -q 20 -f 5 -r 0 -v 0.05 -s 16 - s1 '。在配对端测序的情况下,重复序列被sambamba掩盖。然后使用MethylDackel从对齐的读数中提取CpG位点的甲基化指标。
2、DNA 甲基化指标
使用 mHapTools从 BAM 文件中提取 DNA 甲基化单倍型。随后,生成的 mHap 文件被用作计算各种 DNA 甲基化指标的输入,包括平均甲基化、PDR、CHALM、MHL 和 MCR。PDR 和 CHALM 是根据至少覆盖四个连续 CpGs 的读数计算的,而 MHL 和 MCR 则考虑了通过特定区域的所有读数。为了获得稳健的信号,缺失值被分配给读数少于 10 个的区域。所有 DNA 甲基化指标都是用 mHapSuite(https://github.com/yoyoong/mHapSuite)计算的,它是 mHapTk 的 Java 实现。
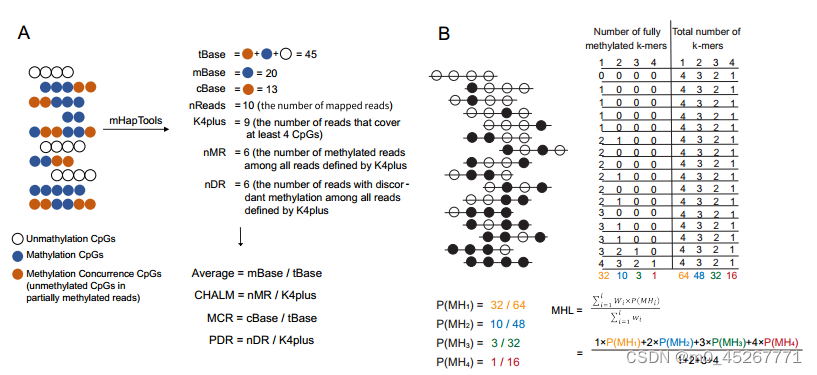

3、TRAmHap 的架构
我们开发了一种名为 TRAmHap 的深度学习模型,用于从 DNA 甲基化数据中预测基因表达谱。TRAmHap 由两个主要部分组成:一个 CNN 模块和一个递归神经网络(RNN)模块。模型的输入是一个三维张量,代表基因组数据被划分为不同的区间和计算特征。CNN 模块使用两个并行卷积层进行特征提取,两个卷积层的核大小分别为 1 × 3 和 1 × 5。两个卷积层的输出被串联起来,形成一个组合特征表示,然后由 RNN 模块处理,该模块旨在使用一系列 LSTM 层识别基因组数据中的模式。然后,RNN 模块的输出通过全连接层生成最终预测结果。TRAmHap 使用 Python 3.8.6 中的 Pytorch(v1.9.0)实现。我们采用leave-one-out strategy(留一法交叉验证) 来预测单个样本的基因表达。训练数据按 8:2 的比例分成训练集和验证集。我们使用均方误差(MSE)作为损失函数,由 Adam 优化器优化,学习率为 5e-4,衰减率为 0.98,每个训练方案中的(β1, β2)=(0.5, 0.998)。我们对模型进行了 20 次历时训练,并将验证集选出的最佳模型保存为结果。此外,该模型的主干可以冻结,并通过迁移学习在不同组织间转移,以提高其性能,特别是在预测不同组织和疾病条件下的基因表达水平时。
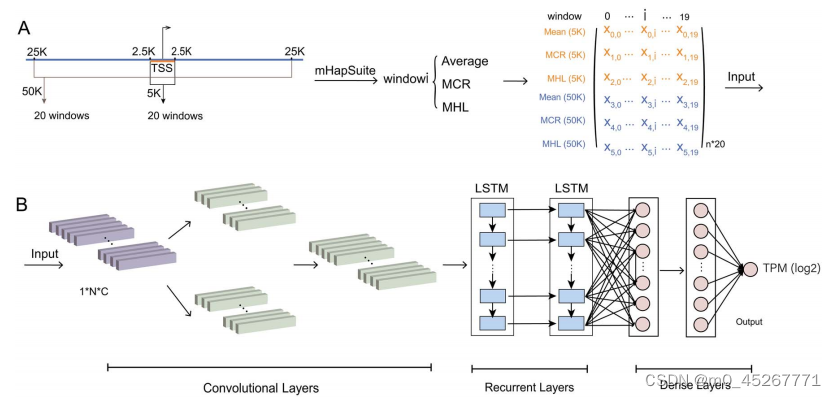
4、DNA 甲基化相关调控潜力
为了评估增强子的影响,我们比较了两个模型的预测性能:含有远端增强子的完整模型(50 kb 和 TSS 周围 5 kb)和不含远端增强子的缩小模型(TSS周围5 kb)。我们将每个基因的预测误差定义为预测值和实际值之间的曼哈顿距离。
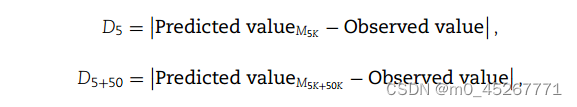
其中,D(5 + 50) 和 D5 分别表示完整模型和缩小模型的预测误差。数值越小,表示预测效果越好。增强子的影响根据 D5 和 D(5 + 50 )之间的差值估算,即
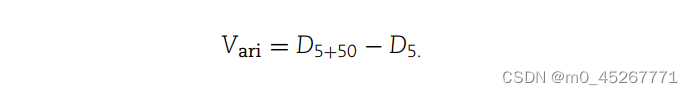
然后根据 Vari 对所有基因进行升序排序。增强组包括在模型中包含 50 kb 区域时预测性能较好的基因,而减弱组包括预测性能较差的基因。无变量组包括 Vari 值接近 0 的基因,即 D(5 + 50) 和 D5 之间没有显著差异。
结果与讨论
1、选择 DNA 甲基化指标作为模型输入
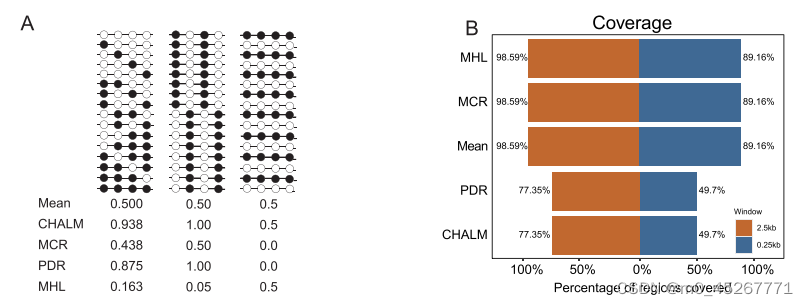
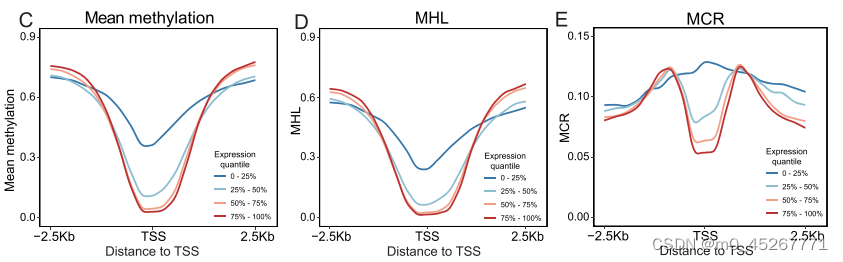
我们的研究建议使用 TSS 周围的 DNA 甲基化模式来预测基因表达。我们关注的是 mHap 水平指标,它不仅测量平均甲基化,还测量读数水平的 DNA 甲基化模式。值得注意的是,对于具有相同平均甲基化水平的区域,mHaps 可以反映出不同的 DNA 甲基化模式。例如,一个平均甲基化为 0.5 的区域可能存在不同的模式,可以用不同的 mHap 水平指标来区分(图 1A)。一些 mHap 水平指标,包括 PDR、CHALM、MCR 和 MHL(补充图 S1),已被证明与基因表达相关。然而,由于 WGBS 数据的读数覆盖率较低,在某些基因组区域可能无法计算 mHap 水平指标。例如,PDR 和 CHALM 只计算覆盖至少四个连续 CpG 位点的测序读数,而当窗口大小设置为 250 bp 时,这些读数只覆盖典型 WGBS 数据集中不到 50% 的区域(图 1B)。即使窗口大小为 2.5 kb,仍有 23% 的数据缺失。另一方面,无论使用何种窗口大小,平均甲基化、MHL 和 MCR 都覆盖了 85% 以上的区域,因此被选为我们模型的可能输入。随后,我们利用ENCODE(ENCBS366XOW)提供的DNA甲基化和基因表达数据,探讨了这些DNA甲基化指标与基因表达之间的关联。不出所料,基因表达量最低的一组在 TSS 附近的平均甲基化程度最高(图 1C)。在 MHL 中也观察到了同样结果(图 1D),而 MCR 在区分四组时更为有效(图 1E,补充图 S2)。因此,平均甲基化、MHL 和 MCR 代表了有效的 DNA 甲基化指标,因此被选为模型输入。
2、TRAmHap 的框架
我们为每个基因定义了两个调控区域,即 TSS 上游和下游分别为 ±2.5 kb 和 ±25 kb。较短的 5 kb 区域覆盖近端启动子,而较长的 50 kb 区域则覆盖启动子和远端增强子。这些区域被进一步划分为 20 个大小相等的窗口。对于每个窗口,我们计算了三个 mHap 级指标,即平均甲基化、MHL 和 MCR,从而得到三个等宽 20 乘 2 的矩阵(图 2A)。利用这些矩阵作为输入,我们开发了基于 DNA 甲基化单倍型预测基因表达的新型深度学习框架 TRAmHap(图 2B)。TRAmHap 可以将一个、两个或所有三个指标作为输入。该模型从一个二维卷积层开始,将输入矩阵特征重塑为一维序列数组,然后是两个并行卷积模块,内核大小分别为 1 × 5 和 1 × 3。通过使用不同大小的核,模型可以提取不同尺度的特征。所有卷积层都通过 ReLU 激活函数与批量归一化层或 maxpooling 层相连。卷积模块根据位置性和不变性的先验知识提取特征。卷积层的输出被连接并输入 LSTM 模块,以提取基于窗口序列的特征。最后,四个全连接层用于拟合和预测基因表达。
3、TRAmHap 可准确预测同一组织内的转录活性
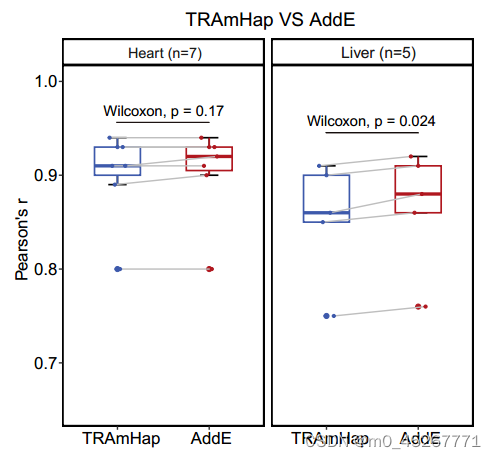
我们使用各种 DNA 甲基化指标作为输入,评估了 TRAmHap 的性能。为此,我们从三种组织类型(包括 7 个心脏样本、7 个前脑样本和 5 个肝脏样本)中筛选出了具有匹配基因表达和 DNA 甲基化图谱的数据集(补充表 S1)。对于每种组织类型,我们都采用了 leave-one-out cross-validation方案,即循环选择一个样本作为测试集,并将其余样本按 8:2 的比例组合成训练集和验证集。我们通过计算测量基因表达和预测基因表达之间的皮尔逊相关系数(PCC)来评估模型的性能。结果表明,在所有三种小鼠组织类型中,基于平均甲基化和MCR的TRAmHap模型优于基于其他指标或所有三个指标的模型,在心脏、前脑和肝脏组织中,PCC中值分别为0.91、0.87和0.86(图3A)。
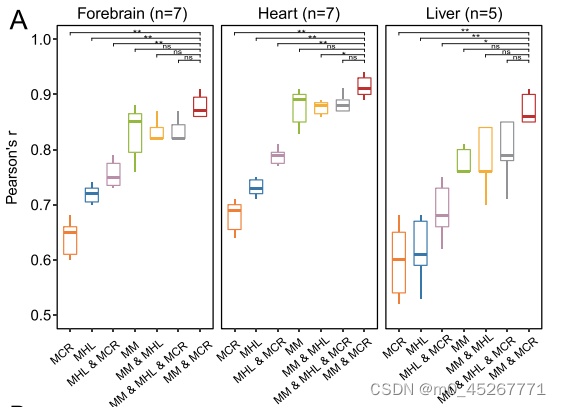
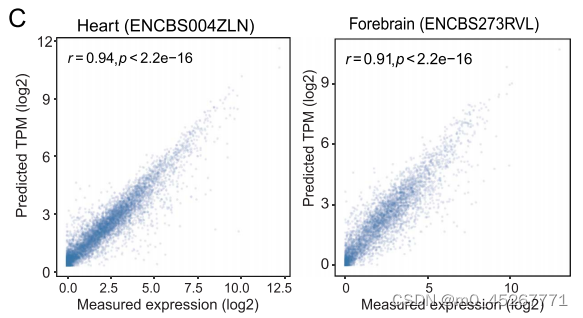
总之,这些研究结果表明,平均甲基化和 MCR 是 TRAmHap 预测基因表达的最佳输入。利用 TRAmHap 模型,观察值和预测值之间的 PCC 为 0.94(图 3C,左侧面板),表明基因表达预测准确。脑组织样本(ENCBS273RVL)也得到了类似的结果(图 3C,右图)。
我们随后评估了将基因组区域划分为不同数量窗口时 TRAmHap 的性能。窗口尺寸越大,窗口数量越少,覆盖窗口的比例越高;窗口尺寸越小,窗口数量越多,覆盖窗口的比例越低。我们要求每个基因至少有 90% 的窗口被覆盖,否则该基因将被排除在进一步分析之外。以人类正常组织数据集为例,当区域被分割成 20 个窗口时,大约有 10,000 个基因被保留下来,而当区域被分割成 30 个窗口时,只有 200 个基因被保留下来(图 3B 左侧面板)。虽然窗口数越少覆盖基因的数量越多,但是20个窗口拥有更好的皮尔逊相关系数(PCC)。在整体预测准确性方面,将区间划分为 20 个窗口的模型优于划分为 10 个窗口的模型和划分为 30 个窗口的模型(图 3B,右图)。

4、TRAmHap 与现有方法的比较
我们将 TRAmHap 与现有的几种通过 DNA 甲基化预测基因表达的方法进行了比较分析。这些方法包括线性回归(LR)、支持向量回归(SVR)、随机森林(RF)和 CNN。上述所有方法都使用了来自三种组织类型的相同数据集:七个心脏组织样本、七个前脑组织样本和五个肝脏组织样本。为确保公平比较,所有方法都在相同的训练集和验证集上进行了评估。对于 SVR 和 RF,我们在广泛的参数组合范围内进行了网格搜索,并在随后的结果中选择了最佳参数进行建模和比较。有关网格搜索的详细信息,请参见补充表格(补充表格 S2)。在所有数据集上,TRAmHap 的表现都优于所有其他方法,具体表现为预测基因表达量与真实基因表达量之间的 PCC(图 4A)。具体来说,TRAmHap 的 PCC 中值为 0.85,通过 DNA 甲基化解释了约 72% 的基因表达变异。相比之下,所有其他方法的中位 PCC 值都低于 0.8,其中经典 CNN 的表现最差,在小鼠心脏、大脑和肝脏组织中的中位 PCC 值分别为 0.47、0.41 和 0.36。与 LR、SVR 和 CNN 相比,RF 的表现更为稳健,但只能解释 50%的基因表达变异(中位数 PCC ≈ 0.74)。
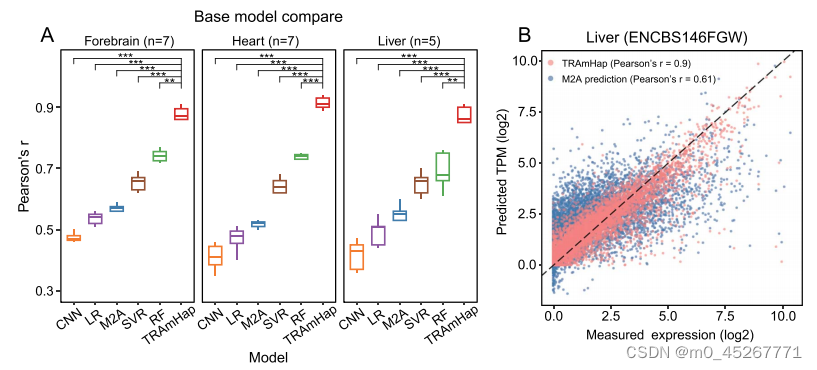
在最近的一项研究中,作者开发了一种名为 MethylationToActivity(M2A)的深度学习框架,通过测量启动子 ±1 kb 区域内 H3K4me3 和 H3K27ac 的富集程度来预测启动子的活性(补充图 S4)。它预测的结果的皮尔逊相关系数(PCC)要优于TRAmHap的深度学习框架。然后,我们在本研究使用的样本上测试了 M2A,将组蛋白修饰信号替换为基因表达作为输出。然而,M2A 预测基因表达的准确性很差,在小鼠心脏、大脑和肝脏组织中的 PCC 中值分别为 0.57、0.52 和 0.55。例如,对于小鼠肝脏样本(ENCBS146FGW),TRAmHap 的 PCC 值为 0.90,而 M2A 的 PCC 值仅为 0.61(图 4B)。这些发现表明,在利用 DNA 甲基化数据预测基因表达方面,TRAmHap 优于基本的机器学习模型和现有的深度学习模型。
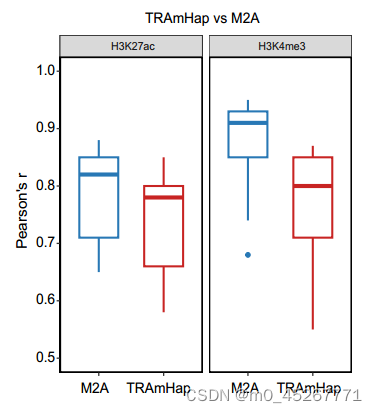
5、TRAmHap 可预测不同组织和疾病条件下的转录活性
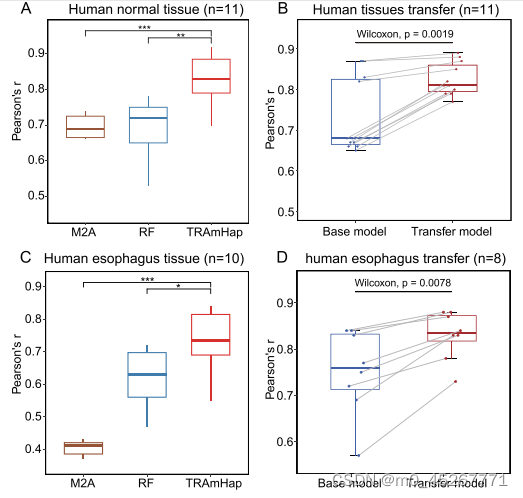
我们进一步探讨了当训练样本和测试样本来自不同组织时,所提出的模型是否仍具有预测能力。举例来说,我们在肺组织样本上测试了我们的方法,使用的是用所有其他组织训练的模型。虽然基因表达的预测准确率略低于组织内预测的准确率,但该模型仍然达到了相当高的准确率(PCC = 0.89)。
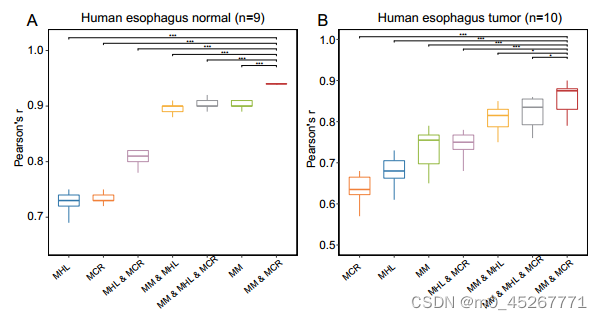
随后,我们通过测试来自相同组织但处于不同疾病状态的样本来评估模型的普适性。我们从 GEO 数据库(GSE149612)中获得了一个数据集,其中包括食管的正常组织样本(n = 9)和癌症组织样本(n = 10)。与之前的结果一致,用正常样本训练的模型能准确预测其他正常样本的基因表达,中位 PCC 为 0.94。同样,用癌症样本训练的模型在预测其他癌症样本的基因表达方面也达到了很高的准确度,中位 PCC 为 0.88(补充图 S5)。值得注意的是,尽管涉及不同的调控机制,用正常样本训练的模型也能合理预测肿瘤样本,中位 PCC 为 0.74。与 RF 和 M2A 相比,TRAmHap 的表现大大优于它们(图 5C)。通过迁移学习,TRAmHap 的性能进一步提高,PCC 中值达到 0.80(图 5D)。
6、TRAmHap 对增强子的利用
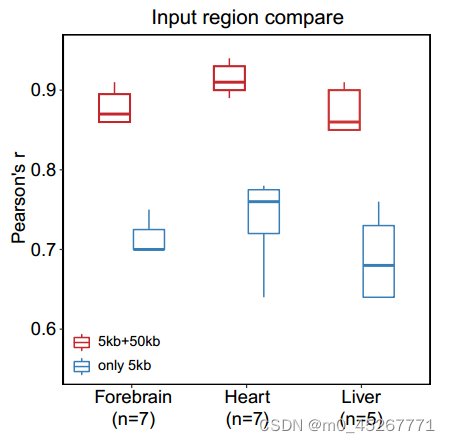
在我们对 TRAmHap 模型的研究中,我们观察到输入间隔为 50 kb 和 5 kb 的模型的性能优于输入间隔仅为 5 kb 的模型,这使得在相同组织内预测效应的皮尔逊相关系数(PCC)显著提高。具体来说,小鼠前脑、肝脏和心脏组织的 PCC 中值分别增加了 0.17、0.16 和 0.16(补充图 S6)。这种改善可能是由于假设较大的区域含有增强子,可以提高模型的性能。为了验证这一假设,我们比较了输入和不输入长范围区域(TSS ± 25 kb)时模型的性能。我们根据预测误差的变化对基因进行了排名,排名靠前的基因预测能力增强,排名靠后的基因预测能力减弱,而处于中间位置的基因没有明显变化。在心脏组织中,我们发现预测增强的前 100 个基因中有 61% 包含已知的心脏特异性增强子(图 7A)。相比之下,前 100 个未受影响的基因中只有 14% 含有已知的增强子。有趣的是,预测结果降低的前 100 个基因中有 50%也含有已知的增强子。
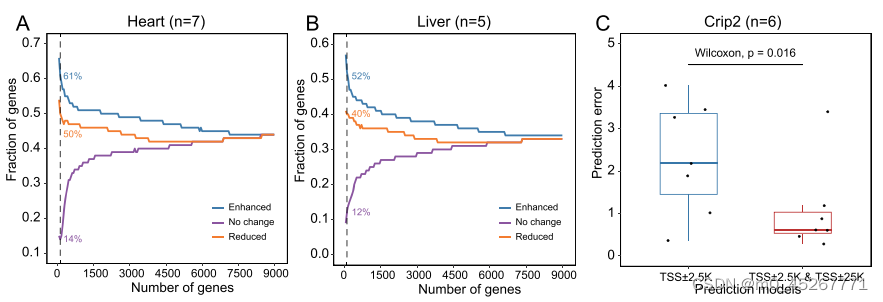
基于上述结果,我们尝试在模型输入中加入组织特异性增强子信息,即每个窗口的平均甲基化、MCR 和增强子信息('1'表示该窗口包含该基因的增强子,'0'表示该窗口不包含该基因的增强子),结果表明加入这些信息后,模型的预测结果略有改善(补充图 S9)。这一结果也再次表明,在预测基因表达时考虑增强子信息有利于提高模型的准确性。
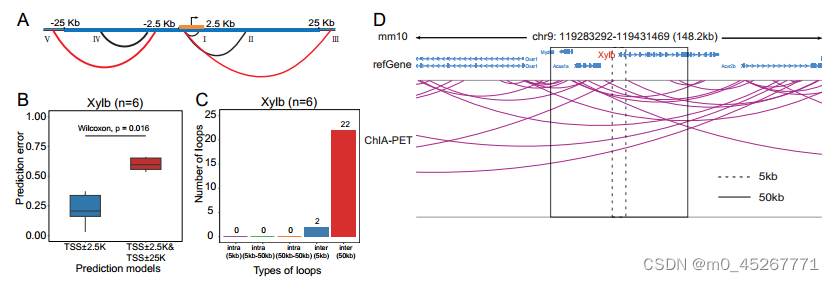
然而,我们观察到,预测结果降低的基因中也含有一小部分增强子,但当模型中加入 50kb 区域时,预测误差增加了。众所周知,增强子远离启动子,通过远端染色质相互作用调节基因表达。在这些情况下预测误差减小的一个可能解释是,一些增强子可能位于相关基因的 50kb 区域内,但却能调控这些基因位点之外的基因(补充图 S10A)。为了验证这一假设,我们比较了预测增强、预测减弱或预测不变的基因组中基因内和基因间联系的比例,这些联系是通过成对标记测序染色质相互作用分析(ChIA-PET)确定的。预测增强的基因组往往包含较高比例的基因内链接和较低比例的基因间链接。有些基因既包含基因内链接,也包含基因间连接,但预测性降低的基因往往以基因间连接为主(图 8C)。
讨论
根据上述观察结果,我们开发了一种名为 TRAmHap 的新型深度学习框架,它可以利用距离 TSS 长达 25 kb 的近端启动子和增强子中 DNA 甲基化单倍型的特征来预测转录活性。根据 DNA 甲基化指标的不同,框架内有多种模型可供选择。传统的平均甲基化提供的是聚集信号,忽略了细胞群的异质性,而 PDR、CHALM、MCR 和 MHL 等 DNA 甲基化指标则考虑了异质性,捕捉到了 DNA 甲基化单倍型的模式。然而,PDR 和 CHALM 计算只考虑至少覆盖四个 CpG 位点的测序读数,导致典型的 30× WGBS 样本在 TSS 周围 5 kb 区域的缺失值超过 50%。因此,我们选择平均甲基化、MCR 和 MHL 作为模型输入。窗口尺寸越大,窗口数量越少,覆盖窗口的比例越高;窗口尺寸越小,窗口数量越多,覆盖窗口的比例越低。我们使用不同的窗口数检验了 TRAmHap 的性能,发现 20 个窗口(覆盖约 9000 个基因)的效果最好。虽然 10 的窗口大小覆盖了超过 10 000 个基因,但 TRAmHap 的性能明显下降。因此,20 的窗口数兼顾了性能和覆盖率。
在组织内预测方面,基于平均甲基化和 MCR 的 TRAmHap 模型优于其他基于 DNA 甲基化特征指标的模型。这一结果表明,DNA 甲基化单倍型的特征有助于预测转录活性。我们认为 TRAmHap 框架还可以进一步完善,加入更多层次,如具有注意机制的层次,以提高噪声容忍度。
结论
在这项研究中,我们开发了一种新的机器学习框架,利用启动子和增强子中 DNA 甲基化的特征来预测基因表达。该模型优于现有模型,可预测不同组织和疾病条件下的基因表达。我们的模型表明,DNA 甲基化特征可用于准确预测基因表达。此外,我们还发现,基因表达不仅取决于TSS附近区域(±2.5 kb)的DNAm,还取决于远端增强子,尤其是当基因内染色质相互作用存在时。

















 1804
1804

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









