







目前,针对群体的研究基本上还是以重测序为主,基于对遗传多样性丰富的自然群体中的个体进行全基因组重测序,研究物种遗传进化多样性,结合准确的目标性状的表型数据及统计方法进行全基因组关联分析,可对动植物复杂农艺性状进行定位,快速获得影响目标性状表型变异的遗传标记或候选基因。
随着表观遗传技术的发展,越来越多的表观技术也应用到群体的研究上,特别是DNA甲基化测序(比如WGBS),研究者们通过获得群体的DNA甲基化数据后进行种群DMR分析,与重测序数据关联分析、EWAS分析、meQTL分析获得影响目标性状表型变异的表观遗传标记以及候选基因。
我们先回顾一下重测序的分析策略。首先拿到大量样本(不同群体)的测序数据,然后利用FastQC进行原始数据的质控和过滤;得到质控结果后,再将过滤后的数据比对到参考基因组上,并进行排序和去重复等处理,利用BWA比对和samtools软件进行格式转化为bam文件;再利用GATK进行SNP和INDEL检测生成VCF文件;用lumpy得到结构变异(structure variants)的信息以及CNVnator分析得到拷贝数变异(Copy Number Variation,CNV)的VCF结果;利用ANNOVAR对SNP/INDEL、SV以及CNV进行注释;接下来构建进化树,PCA分析以及structure分析;得到群体之间的进化关系信息。LD衰减分析;群体选择分析(Tajima’D分析,Fst分析以及ROD分析);针对有表型数据的,可进行GWAS分析;得出性状与SNP/INDEL之间的关联信息;再通过QTL分析精准定位与目标性状相关的遗传标记或候选基因。
接来下我们看一下群体甲基化的分析流程:
01
群体的选择以及样本个数
参考已发表文献,我们可以发现所用群体以自然群体为主,也有一些自交群体。此外,2021年的一篇Science则是以不同物种的DNA甲基化进行后续分析(参见:动物群体甲基化如何讲故事?)。

表1:不同文献中的群体选择以及样本个数。
02
基因组比对
常见的分析是直接将过滤后的数据比对到参考基因组。在有重测序的数据情况下,文献大部分都是比对过滤snp后的参考基因组。
03
计算每个样本的DNA甲基化水平
比对参考基因组后要计算每个样本的全基因组甲基化水平。计算公式如下:C位点的甲基化水平=100*支持甲基化的reads/(支持甲基化的reads+支持非甲基化的reads)。
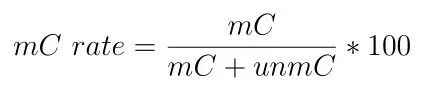
甲基化 C 碱基在基因组上的分布包含三种形式(CG,CHG 和 CHH,其中 H 代表 A 或T 或 C 碱基)。利用 cgmaptools[6] 软件(version: 0.1.1)统计各种类型的 C 碱基的甲基化水平的比例分布,在一定程度上反映了特定物种的全基因组DNA甲基化修饰特征,并且可计算不同甲基化位点的数量和比例。
04
DMR分析:可分析高频可变区域以及低频可变区域
-
筛选条件:
(1) 判定 C 选定区间,保证至少有 5 个 C 碱基并且所有 C 碱基深度大于 5x,这些区间最长1000bp 长度,两个短于1000bp的选定区域距离不小于 200bp;
(2)根据两个样本的选定区间判定差异DMR区间,阈值为Pvalue小于等于 0.001,DMR 水平大于等于 0.2。
DMR的筛选条件文献也是有不同的,可以根据测序得到的结果进行调整。
针对不同种群可以绘制不同种群的整体甲基化水平、差异DMRvenn图、热图。看不同种群之间是否存在甲基化整体水平的差异,以及DMR区域。DMR在基因不同区域,Exon、Intron、TE、Intergenic情况展示。
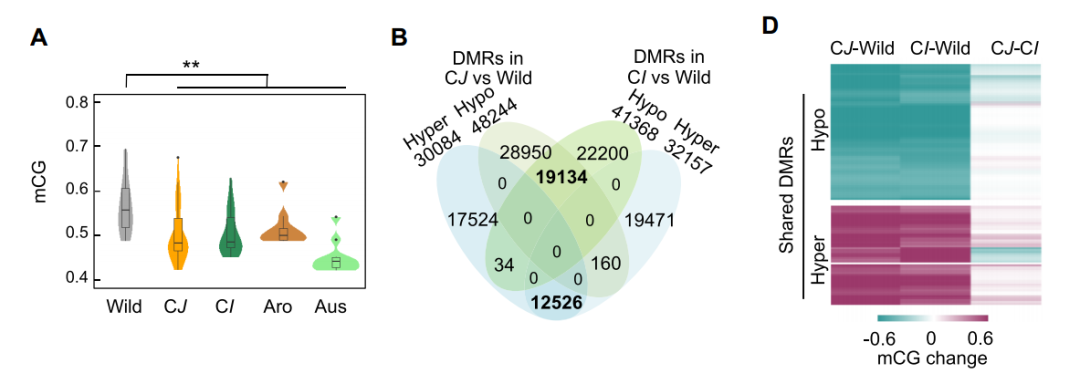
图1:A.不同种群的甲基化水平;B.DMR的venn;D.DMR热图。
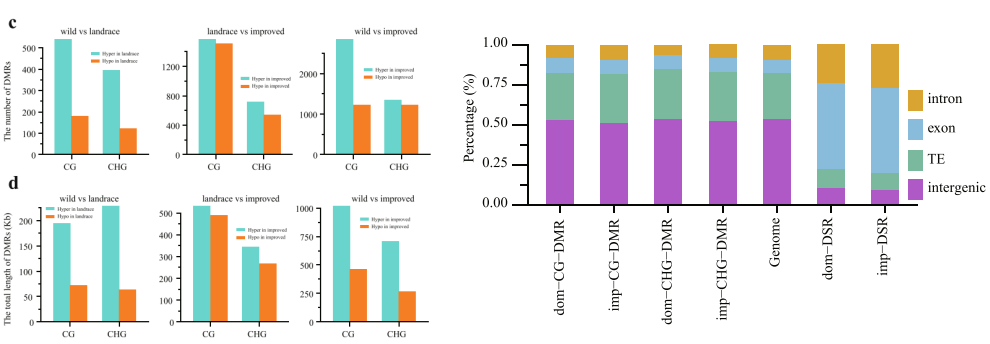
图2:DMR的数量和长度统计以及DMR在不同元件的分布。
05
DMR注释,GO和KEGG富集分析
DMR 区域中点与基因或基因的 Promoter 区域(TSS 上游 2kb)有交集,认为与该基因有关联;然后,针对关联基因进行GO和KEGG富集分析。
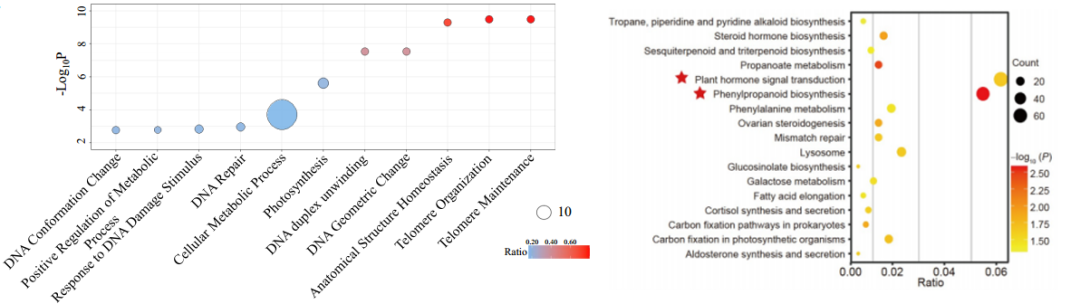
图3:DMR关联基因的GO和KEGG富集分析
06
WGBS和SNP关联分析以及PCA分析
使用SNP计算的成对亲缘关系与基于CG甲基化水平的亲缘关系高度相关(图4B),这表明DNA甲基化的变化可以概括不品种之间的遗传关系。此外,利用CG甲基化变异的主成分分析(PCA)成功地将品种划分为不同的亚群(图4C),目前看到的文献结果与基于SNP的分类一致。
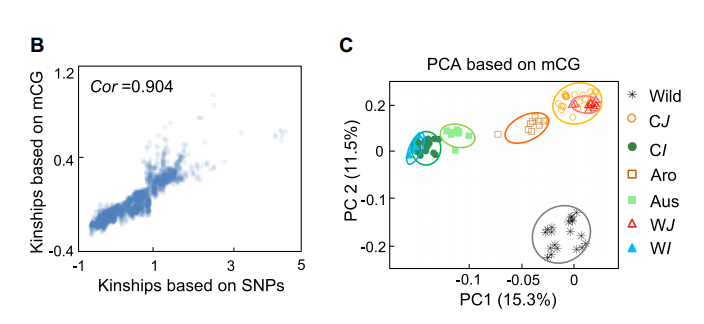
图4:B.通过SNP或mCGS水平计算样本之间亲缘关系;C.基于CG甲基化水平的所有水稻品种主成分分析
07
EWAS分析
GWAS(基因组关联研究)是一种用来找新基因和基因区域的方法,可以帮助我们定位复杂疾病/表型的关键基因。不过GWAS只能在遗传信息层面上解读复杂疾病/表型,无法涉及表观遗传学。因此,新的方法出现了,名为表观基因组关联分析(EWAS),EWAS将表观遗传的变异和复杂疾病/表型联系起来,通过研究表观遗传学来解读复杂疾病/表型的原因,找到与疾病/表型相关的表观遗传学变异位点。
-
7.1 EWAS可以检测受环境因素影响的新的调控机制
EWAS可以将在实验组全基因组范围内检测出的甲基化变异位点与对照进行比较,找出所有甲基化位点的变异频率,同时还可以鉴定新的与疾病/表型的甲基化位点。
-
7.2 EWAS利用探究DNA序列变异和DNA甲基化之间的关系。
大部分GWAS显著关联位点落在基因组非编码区,其如何通过基因或者通路影响表型很难被阐述,一种可能得解释是,这些易感位点通过调节特定区域的甲基化水平,从而改变个性复杂形状。如果某个位点即对负责形状有影响,又对甲基化水平有影响,那么该位点就很有可能符合上述解释。共定位分析(Collocalization)正是试图找出这些“共定位”位点。共定位分析方法,属于Post-GWAS的一项重要工作,eQTL和mQTL是EWAS常用的共定位方法,旨在GWAS结果的基础上鉴定与表型相关的eQTL和mQTL位点。SMR利用GWAS的summary数据和表达数量性状基因座(eQTL)的数据,采用SMR和HEIDI方法,以测试基因表达水平与感兴趣的复杂性状之间的多效性关联。
目前,主要用到软件EWAS2.0(Xu et al., 2018a)进行分析。EWAS2.0软件可以进行:(i)全表观基因组单标记关联研究;(ii)表观基因组甲基化单倍型(meplotype)关联研究和(iii)表观基因组关联荟萃分析。
对于物理上彼此接近的多个DNA甲基化位点,这些位点之间存在表观等位基因的非随机关联,称之为甲基化不平衡(methylation disequilibrium, MD)。EWAS2.0可以计算MD系数识别MD块,并使用Excoffier等人的最大似然估计方法估计meplotype(染色体上一组特定的外显等位基因)的频率。对于病例/对照数据,EWAS2.0可以扫描整个表观基因组,识别疾病相关的meplotype(计算卡方、p值、奇比和95%保密区间)。EWAS2.0可以扫描整个表观基因组,识别疾病相关meplotype(计算卡方、p值、奇比和95%保密区间)。首先需要进行单个SMP分析后进行meplotype分析,以确定与疾病/表型相关的一些SMP位点的组合。EWAS2.0使用Cochran’s q统计量检验个体研究之间的异质性。
EWAS的可视化结果图和GWAS类似,曼哈顿图、QQ图和LD-Block图。下面是文献的展示结果:
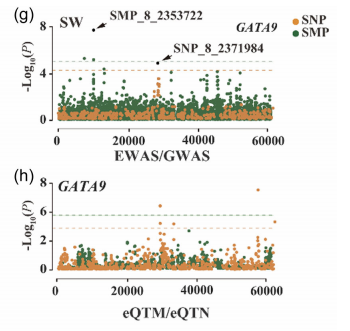
图5:基于SMPs和SNP关联分析的曼哈顿图以及Cis调控SMPs和SNPs在基因表达中的关联结果
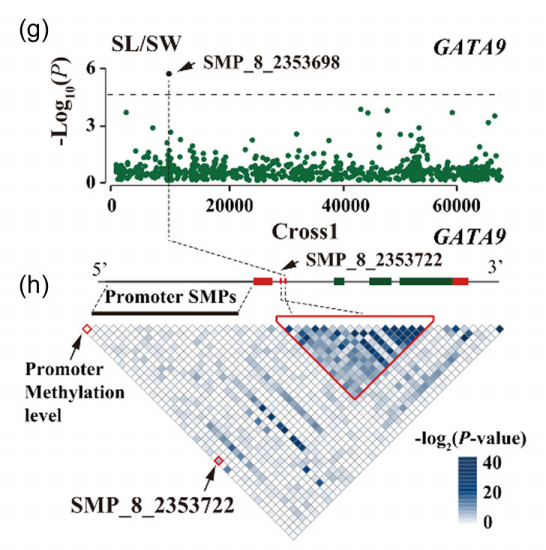
图6:(g)曼哈顿图显示Cross1中GATA9的SL/SW比值关联结果。(h)跨GATA9的SMPs之间的连锁不平衡和由单侧排列检验鉴定的显著位点组成的稳定连锁(p < 0.001)。
其中,番茄的研究结果还加入了代谢组的分析结果,展示图如下:
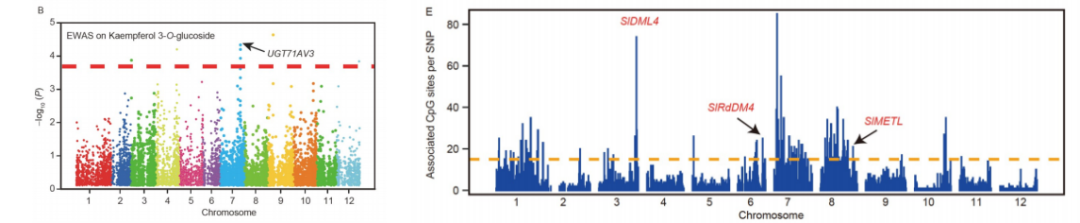
图7:山奈酚3- o -葡萄糖苷的EWAS曼哈顿图。meQTL信号在番茄基因组中的分布。
08
WGCNA分析
DNA甲基化数据也可以构建WGCNA网络。使用WGCNA方法,将具有相似甲基化模式的CpG位点组成共甲基化模块,并用“模块特征基因”来总结这些模块的甲基化特征。
传统上,WGCNA应用于转录组数据,并使用无监督聚类方法将共表达基因分配到模块7。在“DNA Methylation Networks Underlying Mammalian Traits”这项研究中,作者使用WGCNA方法来定义哺乳动物样本中共甲基化CpGs的模块。首先,使用带符号矩阵的软阈值功率(调优值= 12)将邻接矩阵(cpg之间的相关性)转换为无标度网络。将结果转化为拓扑重叠矩阵(TOM)和1-TOM距离度量(不相似度),用于数据的分层聚类。使用动态树切算法对树进行修剪,以分配包含至少30个cpg的模块。基于奇异值分解方法,计算模块特征基因(MEs)为每个模块单个变量所能表示的模型方差的最大量。eutherian网络(Net 1)中的特征基因解释了24-63%(平均= 43%)的特征基因。每个模块中甲基化数据的差异(表S3)。基于特征基因连通性(eigengene connectivity, kME)定义了各模块的hub CpGs。采用多元线性回归模型对不同性状的模特征基因进行关联分析。使用WGCNA包中的matchLabels()函数对两个网络中的模块颜色进行匹配。利用WGCNA R包中的“modulePreservation”R函数,以灵长类动物为参照进行比较,估计各网络的模块保存情况。

总之,群体甲基化分析策略为首先选择合适的群体,然后拿到WGBS数据后比对参考基因组,再进行每个样本甲基化水平,以及不同群体的DMR,然后与重测序数据进行关联分析,PCA分析,以及EWAS分析和meQTL定位,还可以加入WGCNA分析,最终得到表观遗传标记对群体进化/驯化或者人类疾病的影响。
-
参考文献:
[1] Haghani A, Li CZ, Robeck TR, et.al DNA methylation networks underlying mammalian traits. Science.PMID: 37561875.
[2] Xu J, Zhao L, et.al EWAS: epigenome-wide association study software 2.0 PMID: 29566144;
[3] Wang Z, Xia A, Wang Q, Cui Z, Lu M, Ye Y, Wang Y, He Y. Natural polymorphisms in ZMET2 encoding a DNA methyltransferase modulate the number of husk layers in maize. Plant Physiol. 2024 Mar 2:kiae113. doi: 10.1093/plphys/kiae113. Epub ahead of print. PMID: 38431291.
[4] Cao S, Chen K, Lu K, Chen S, Zhang X, Shen C, Zhu S, Niu Y, Fan L, Chen ZJ, Xu J, Song Q. Asymmetric variation in DNA methylation during domestication and de-domestication of rice. Plant Cell. 2023 Sep 1;35(9):3429-3443. doi: 10.1093/plcell/koad160. PMID: 37279583; PMCID: PMC10473196.
[5] Song B, Yu J, Li X, Li J, Fan J, Liu H, Wei W, Zhang L, Gu K, Liu D, Zhao K, Wu J. Increased DNA methylation contributes to the early ripening of pear fruits during domestication and improvement. Genome Biol. 2024 Apr 5;25(1):87. doi: 10.1186/s13059-024-03220-y. PMID: 38581061; PMCID: PMC10996114.
[6] Guo H, Cao P, Wang C, Lai J, Deng Y, Li C, Hao Y, Wu Z, Chen R, Qiang Q, Fernie AR, Yang J, Wang S. Population analysis reveals the roles of DNA methylation in tomato domestication and metabolic diversity. Sci China Life Sci. 2023 Aug;66(8):1888-1902. doi: 10.1007/s11427-022-2299-5. Epub 2023 Mar 23. PMID: 36971992.
[7] Zhou J, Xiao L, Huang R, Song F, Li L, Li P, Fang Y, Lu W, Lv C, Quan M, Zhang D, Du Q. Local diversity of drought resistance and resilience in Populus tomentosa correlates with the variation of DNA methylation. Plant Cell Environ. 2023 Feb;46(2):479-497. doi: 10.1111/pce.14490. Epub 2022 Nov 26. PMID: 36385613.
[8] Shen Y, Zhang J, Liu Y, Liu S, Liu Z, Duan Z, Wang Z, Zhu B, Guo YL, Tian Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018 Sep 10;19(1):128. doi: 10.1186/s13059-018-1516-z. PMID: 30201012; PMCID: PMC6130073.
[9] Xu J, Chen G, Hermanson PJ, Xu Q, Sun C, Chen W, Kan Q, Li M, Cri

















 5160
5160

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









