






2024年5 月 9 日, Theis Lab 和 Satija Lab(团队详情在文末)在《 Cell || Perspective》期刊上发表了一篇题为“The future of rapid and automated single-cell data analysis using reference mapping”的综述论文,其中指出随着单细胞数据集数量的迅速增长,将新数据映射到精心策划的参考图谱的工作流程为生物学界提供了巨大的希望。在此观点中,作者讨论了单细胞参考映射算法面临的关键计算挑战与机遇。同时探讨了映射算法如何能够整合跨越疾病状态、分子模式、遗传扰动及不同物种的多样化数据集,并最终取代手动且费力的无监督聚类流程。
简介
参考数据集(框1)和映射算法正在改变单细胞测序数据集的分析工作流程。这与第一个人类基因组图谱构建所带来的类似趋势相呼应。基于参考的分析将数据解释从无监督领域转变为有监督领域,使得从多个先前实验中积累的信息能够帮助解释新数据。在分析基因组序列数据时,参考图谱的存在确保了每个新实验无需从数据本身重新组装基因组,极大地简化了分析工作流程,并降低了对读长和数据质量的要求。同样,对于单细胞分析,高效的参考映射工作流程可以用自动映射和注释取代手动、费力且主观的无监督聚类和标记任务。
框1 术语表
多模态参考(Multimodal reference):使用不止一种模态(例如,RNA 和 ATAC)构建的参考图谱。
多组学(Multimodal omics):能够从同一样本中捕获多种数据类型的技术。
有监督学习与无监督学习(Supervised vs. unsupervised learning):在这种背景下,数据整合时利用参考数据和查询数据集中的细胞类型标签(有监督),与方法无法获取这些标签的情况(无监督)相对。
主成分分析(Principal-component analysis,PCA):一种用于降低数据集维度的线性降维技术。
低维表示(Low-dimensional representation):在此背景下,指应用转换(如 PCA)后数据集的降维空间。
层次分类器(Hierarchical classifier):一种机器学习模型,将数据组织和分类为多个层次或嵌套类别的层级结构,允许进行结构化和精细的分类方法。
深度生成模型(Deep generative models):一类人工智能算法,使用深度神经网络学习和生成新的数据样本,能够在图像、文本或音频等不同领域创建新颖且逼真的输出。
异常值检测(Out-of-distribution detection):一项机器学习任务,专注于识别与训练期间学习到的模式显著不同的实例或数据点,帮助模型识别和标记位于已知分布之外的输入,从而增强在现实应用中的稳健性和可靠性。
多示例学习(Multi-instance learning,MIL):一种机器学习范式,其中训练数据被组织成 “包”,每个包包含多个实例(示例)。模型的任务是在包级别进行预测,虽然为包提供了标签,但每个包内特定实例级别的标签是不确定或未知的。这种方法常用于只有部分标签信息可用的场景,适用于图像分类、药物发现和异常检测等任务。
动态模型(Dynamical models):捕捉系统随时间变化的行为和演化的数学表示。这些模型基于一组微分方程或迭代规则描述变量如何随时间变化,能够在物理、生物、经济和工程等各个领域模拟和预测系统动态。
变分自编码器(Variational autoencoders,VAEs):机器学习中的一类生成模型,结合了自编码器和变分推断的元素。VAEs 旨在学习输入数据与潜在空间之间的概率映射,从而能够生成新的数据点。编码器网络将输入数据映射到潜在空间的概率分布,解码器网络从该分布中采样生成数据。VAEs 常用于生成新的数据样本、数据压缩和无监督学习等任务。
流形学习(Manifold learning):机器学习和数据分析中的一组技术,专注于在低维空间中捕捉高维数据的潜在结构或流形。其目标是以保留数据潜在几何关系的方式表示复杂数据。
潜在空间(Latent space):在机器学习背景下,数据表示被学习和编码的低维空间。它是不同模型架构(包括自编码器和生成模型,如变分自编码器和生成对抗网络)中的一个关键概念。
参考图谱(Reference atlas):经过广泛注释和整理的单细胞数据,全面展示特定组织或样本的细胞异质性,作为细胞和分子特征的详细地图。
标签转移(Label transfer):将来自注释良好的参考图谱的标签投影到新生成的查询数据集上。
因组序列参考映射(Reference mapping for genome sequence):将 DNA/RNA 测序短读段与参考基因组比对,以获得读段的基因组身份。
单细胞参考映射(Single-cell reference mapping):将遗传图谱(如单个细胞的转录组)与参考图谱比对,以获得单细胞水平的注释。
单细胞 RNA 测序(Single-cell RNA sequencing):一种用于分析单个细胞基因表达谱的测序技术。
单细胞 ATAC 测序(Single-cell ATAC sequencing):一种用于分析单个细胞内开放染色质区域的测序技术。
跨模态映射(Cross-modality mapping):一种特殊类型的单细胞参考映射,其中查询和参考属于两种不同的模态,例如将 scATAC 图谱映射到 scRNA 参考上。
典型相关分析(Canonical correlation analysis):一种统计方法,用于理解两个数据集之间的关系,捕捉共享方差并识别相关模式。
非负矩阵分解(Non-negative matrix factorization):一种算法,将高维数据分解为低维表示,确保分解后的矩阵所有成分均为非负。
对抗对齐(Adversarial alignment):一种算法,通过减少批次效应和其他混杂变异来协调来自不同来源或平台的数据集。
通过测序进行转录组和表位的细胞索引(Cellular indexing of transcriptomes and epitopes by sequencing,CITE-seq):一种多模态测序技术,能够在单个细胞中同时测量蛋白质和 RNA。
SHARE-seq、SNARE-seq 和 10x multiple:三种不同的测序技术,能够在单个细胞中同时测量开放染色质区域和 RNA。
多组学桥梁(Multi-omics bridge):用于跨模态映射的单细胞多组学数据集,作为查询和参考之间的桥梁。
配对标签(Paired-tag):通过测序对单个细胞进行 RNA 表达和靶向标签化 DNA 的平行分析(Paired-Tag),是一种能够在单个细胞中同时分析不同组蛋白修饰和转录组的测序技术。
多模态变分自编码器(Multimodal variational autoencoders):一种变分自编码器,用于整合多种模态的数据并学习单个联合潜在表示。
代表性细胞采样(Representative cells sketching):一种从整个数据集中采样细胞子集的算法。采样的细胞应能有效保留完整数据集中的细胞异质性和基因表达协方差。
元细胞(Metacell):一种基于细胞遗传分子相似性将同质细胞分组的计算概念,用于代表不同的细胞类型或状态。
跨物种映射(Cross-species mapping):一种单细胞参考映射类型,其中查询和参考来自两个不同物种,用于从进化关系角度理解保守和多样化的细胞类型及基因程序。
蛋白质语言模型进化尺度建模 2(Protein language model evolutionary scale modeling 2,ESM2):一种基于变压器的语言模型,旨在根据氨基酸序列预测蛋白质结构和功能。
基因组坐标转换(Genome liftover):将基因组坐标信息从一个基因组组装转换到另一个,使得能够比较不同版本或不同物种参考基因组的基因组数据。
基因组序列分析中参考映射的广泛应用也为单细胞测序中的类似潜在应用提供了思路。人类基因组参考使得数百万个体的基因组数据能够被映射,将数据置于一个标准化空间中以进行比较分析(图1A)并识别遗传变异。基因组参考还可以作为多种数据类型和模态的支架,包括染色质免疫沉淀测序(ChIP-seq)和高通量染色体构象捕获(Hi-C)技术等表观基因组分析技术。此外,探索多物种基因组参考之间的差异是一种强大的进化和比较基因组学技术。单细胞测序在这些应用中都有类似之处,这凸显了参考映射算法处理多种数据类型(不仅仅是单细胞 RNA 测序)的潜力,如空间分子图谱。然而,基因组分析也揭示了参考映射方法所面临的挑战,单细胞工具的用户也开始遇到这些挑战。这些挑战有两方面,一方面是技术挑战,如将具有结构变异的基因组映射到健康参考的能力,另外一方面数据驱动的挑战,如需要根据新可用数据不断更新参考。
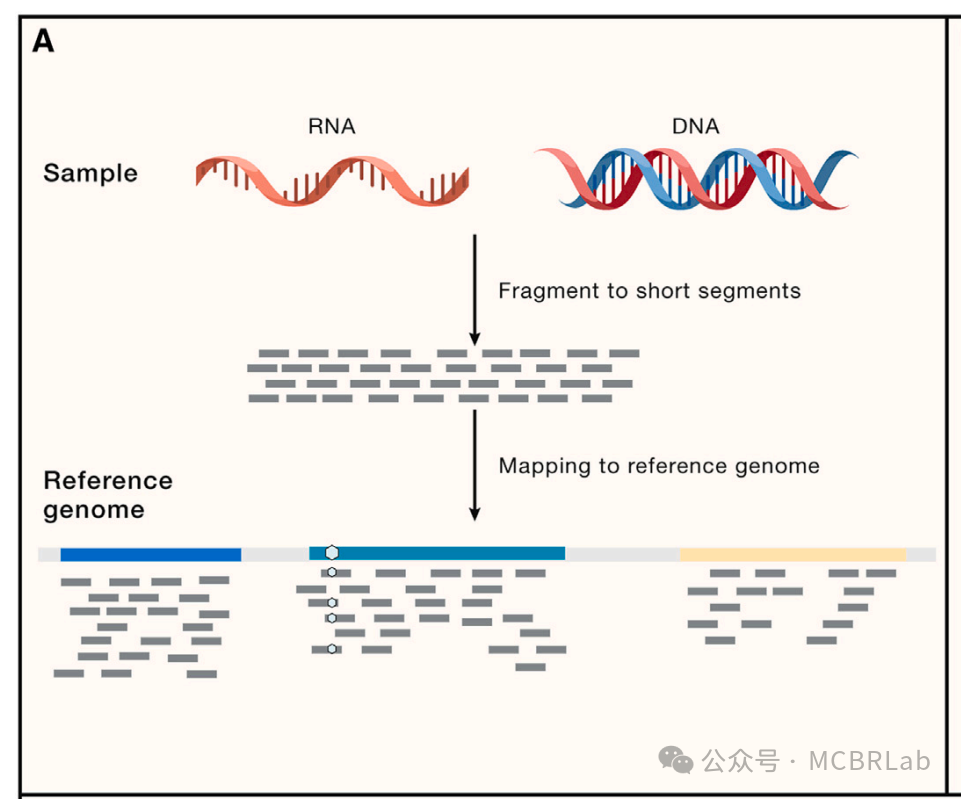
出于这些原因,人们对开发计算方法来组装单细胞参考数据集并将新数据集映射到其上产生了浓厚兴趣。流行的技术涵盖了多种方法,包括基于降维的统计方法、基于机器学习的离散化分类技术和深度人工神经网络。这些技术进展使得能够构建人类器官(如肺、扁桃体和大脑)的单细胞图谱,并使研究人员能够通过整合来自多个研究的数据来研究疾病和发育。这些工具越来越多地与多个合作项目(包括人类细胞图谱和人类生物分子图谱项目)已经组装的参考数据集集合相结合。虽然最常见的分析任务是自动细胞注释(或 “标签转移”),但参考映射工作流程也可以转移连续数据源,包括发育轨迹和其他细胞模态。该领域的计算开发仍在继续,新方法在稳健性、准确性和可扩展性方面不断改进。
在本文中,作者首先回顾单细胞参考映射领域令人兴奋的可能性以及紧迫的挑战。作者还探讨了参考映射工作流程所能涵盖的广泛应用范围以及可以映射的不同查询数据集。这些包括受扰动的细胞(反映疾病状态以及生化或药物诱导的扰动)、跨物种和进化分析以及包含细胞位置和形态信息的空间分辨数据集。最后,作者探讨了在动态领域中构建权威参考所面临的固有挑战,并主张开源图谱能够在新数据可用时快速可靠地更新参考。
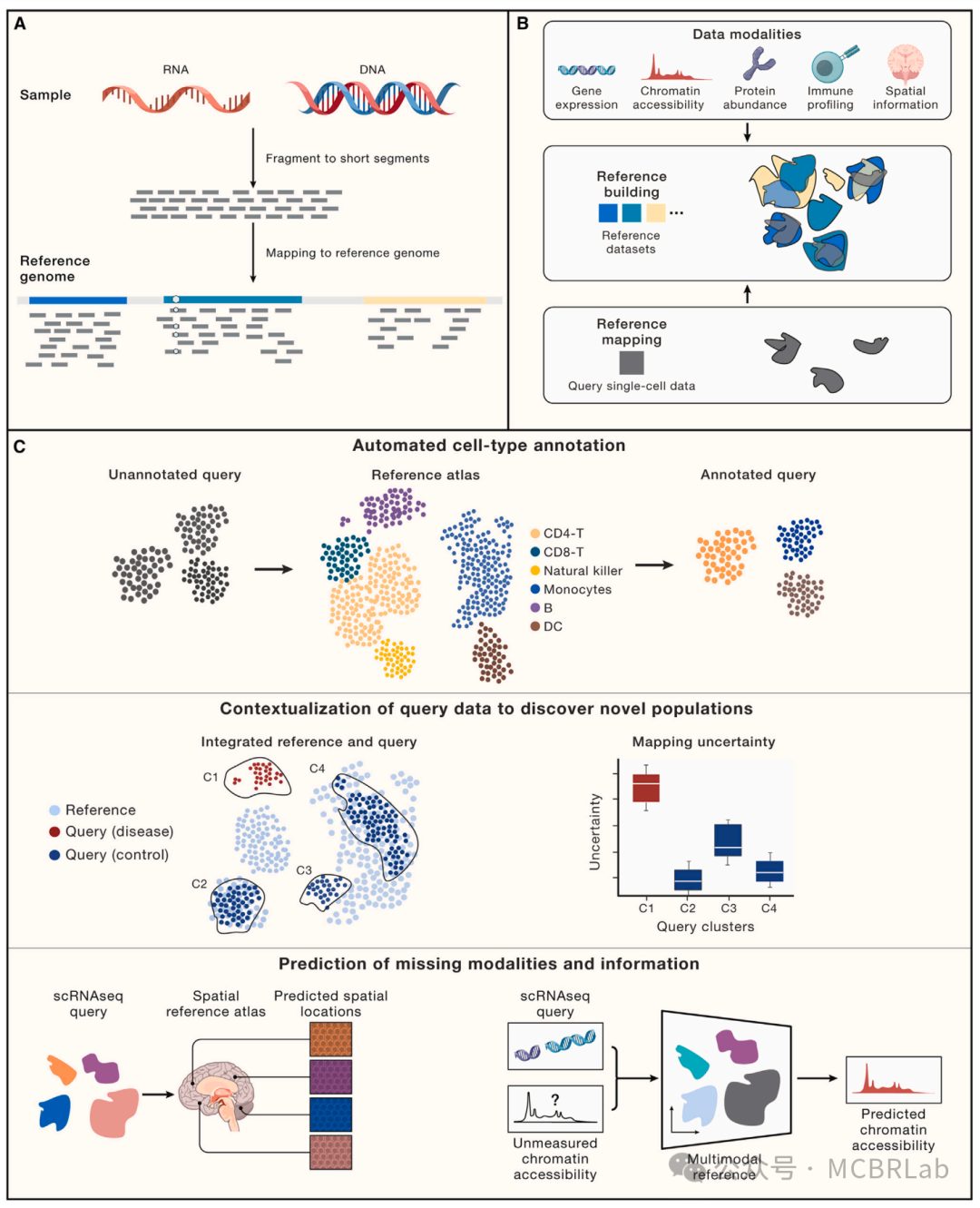
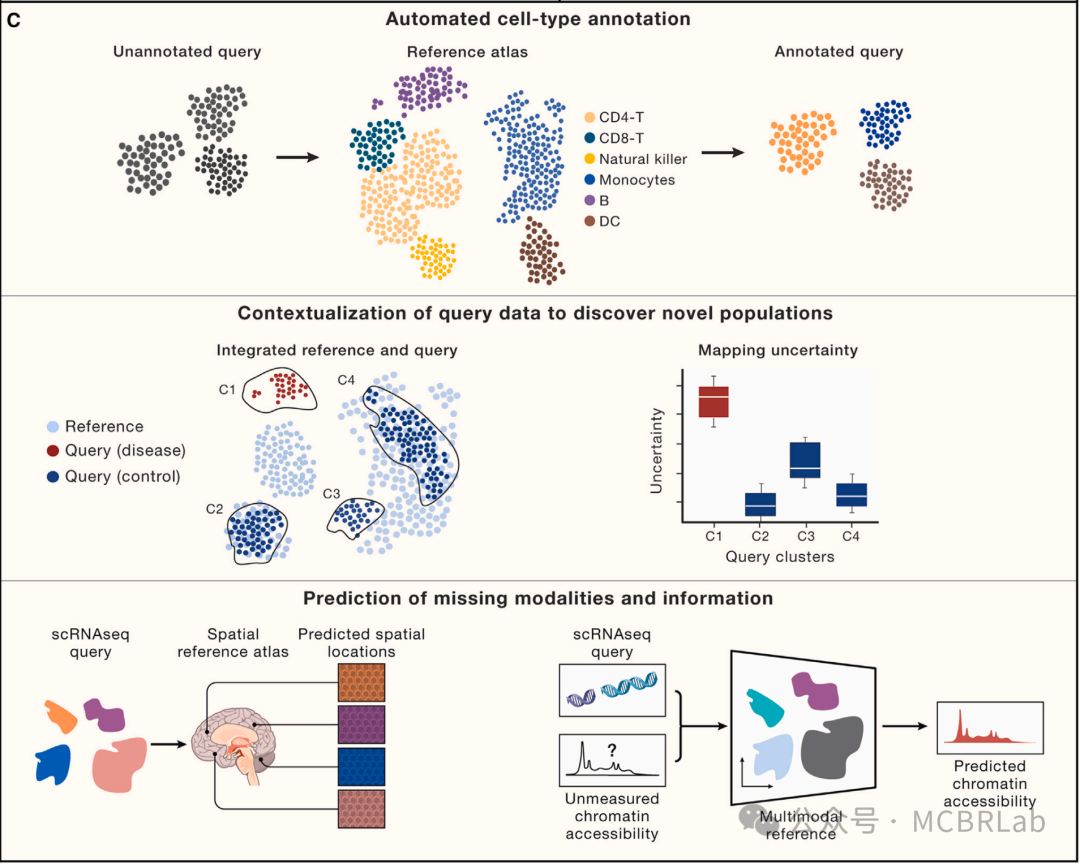
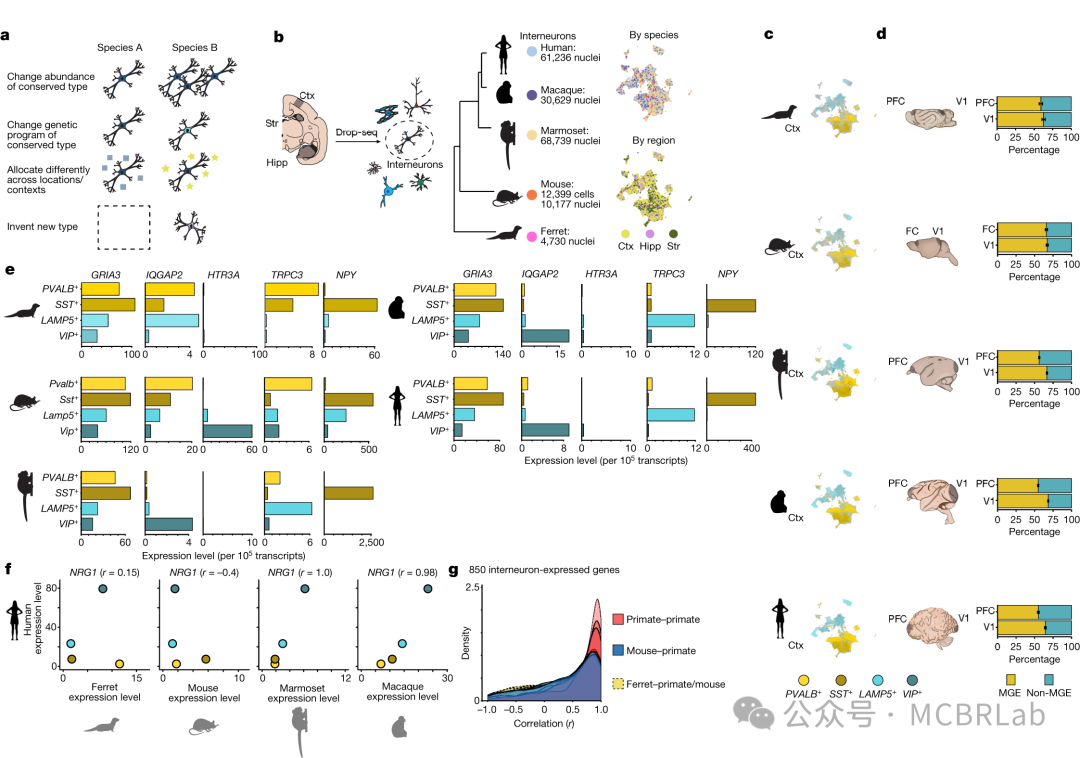
图 1. 使用参考映射对单细胞数据进行自动分析。(A) 使用参考映射器将 RNA 或 DNA 短读段映射到参考基因组,作为计算成本高昂的从头参考组装的替代方法。(B) 单细胞参考的组装——类似于参考基因组——通过使用参考映射算法将新生成的查询数据集映射到参考中,从而实现对其的自动分析。(C) 单细胞参考映射的应用包括查询数据的自动细胞类型注释(第一行)、分析单细胞扰动,如疾病状态或查询数据中要填补的缺失扰动(第二行)、为查询数据填补连续信息,如使用空间图谱填补scRNA-seq的空间位置或使用包括scRNA-seq和scATAC -seq的多模态参考填补查询数据的染色质可及性(第三行)。
参考映射工作流程
将新生成的生物“查询”数据映射到精心整理的参考数据的概念早在单细胞基因组学之前就已存在。例如,基因组参考组装算法计算密集,需要高质量的长读长数据,并且通常涉及大量的手动整理。相比之下,读映射算法高效,与短读长数据兼容且完全自动化。将新的查询数据集映射到已建立的参考而不是对每个数据集进行从头组装和注释具有变革性的影响,并极大地改善了分析数据工作流程。此外,基因组参考可以作为一个单一的支架,以一致地解释和比较来自多个样本的数据。这使得能够深入分析遗传变异,以及跨多种功能基因组学技术(包括ChIP-seq、Hi-C和RNA-seq)进行综合分析。
最近单细胞测序技术的发展导致了高质量的单细胞组织、器官甚至整个物种 “图谱” 的出现。与基因组组装一样,这些图谱的构建通常费力且计算密集,需要手动整理和注释。参考数据通常包含一个或多个模态和元数据的多个单细胞数据集(图 1B),通常包含多达数千种细胞类型及其所在位置信息。单细胞数据整合和参考构建的分析策略和挑战之前已经被综述和比较过。简要地说,单细胞参考数据集通常至少由两个部分组成。第一部分是数据转换,它将数据测量值投影到低维空间。这种转换可以包括多个线性或非线性步骤,通常旨在通过将处于相似生物学状态的细胞放置在相似位置来促进数据整合,即使它们来自不同的数据集。理想情况下,这样的转换应该能够整合跨不同非重叠特征(如基因、峰)的多个数据视图,同时校正来自不同测序协议和环境的技术变异,也称为 “批次效应”。在数据整合的总体主题下,开发方法已经取得了实质性进展,解决了单模态中的批次校正或多模态整合问题,作者将在后面详细讨论。参考的第二部分是元数据的手动分配,通常是为数据集中的每个细胞提供的一组注释,它可以可选地符合已建立的细胞本体并呈现出层次结构。
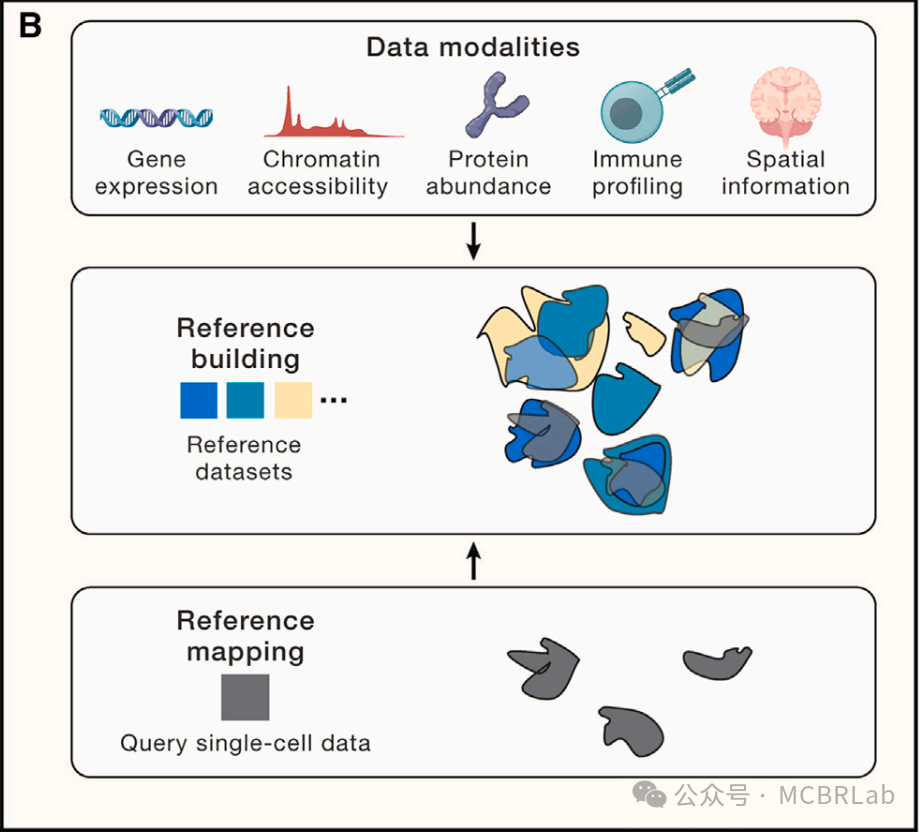
将新样本或查询数据集映射到参考上的不同分析技术也往往遵循一个共同的策略。首先,将在组装参考数据集时学习到的相同数据转换应用于查询。这将查询细胞投影到参考定义的空间中,并有效地整合两个数据集。然后可以利用相邻关系将离散或连续信息基于最相似的参考数据点转移到查询细胞上。虽然这些技术的准确性取决于参考定义的转换和注释的质量,但有监督的映射与无监督分析相比具有显著优势。这些优势包括在有噪声和稀疏查询数据的情况下更高质量的注释、对罕见或分子上细微的细胞状态的改进检测、与不需要手动步骤或参数调整的全自动工作流程的兼容性,以及在速度和内存要求方面的显著改进。最重要的是,参考映射提供了无需重新聚类和重新注释即可解释查询数据集的能力(图1C)。
参考映射方法可以根据为组装参考而学习的转换类型进行分类。第一组方法使用统计方法及其变体学习数据转换。例如,Seurat 使用参考低维表示(如单模态主成分分析 [PCA] 或多模态监督 PCA)投影和基于锚点的整合将查询细胞映射到参考上。同样,Symphony学习一种低维转换(如PCA),其中细胞被软分配到代表不同细胞状态的簇中以构建参考模型。或者,scArches利用概率神经网络学习数据的非线性转换,同时校正数据集之间的技术效应。一旦学习了初始参考转换,就将其应用于查询数据集,根据上一节概述的标准将它们映射到参考上。

无论选择哪种特定的计算方法进行参考映射,这些工作流程都有可能支持广泛的应用。虽然将来自健康个体的数据集映射到健康参考图谱是可能的,但使用健康图谱解释患病样本是非常有必要的,但也伴随着明显的挑战。同样,虽然大多数映射方法是为scRNA-seq数据集量身定制的,但一个关键的计算挑战是映射来自不同数据类型的数据,包括替代模态和空间图谱。最后,参考图谱可能能够对来自不同物种的数据集进行有力的解释,以探索进化变化,或者对来自基因扰动的样本进行解释,以重建分子网络。作者将在下面讨论这些潜在应用及其相关的挑战和机遇(图1C)。
*通过在健康参考中对疾病进行情境化来识别疾病状态*
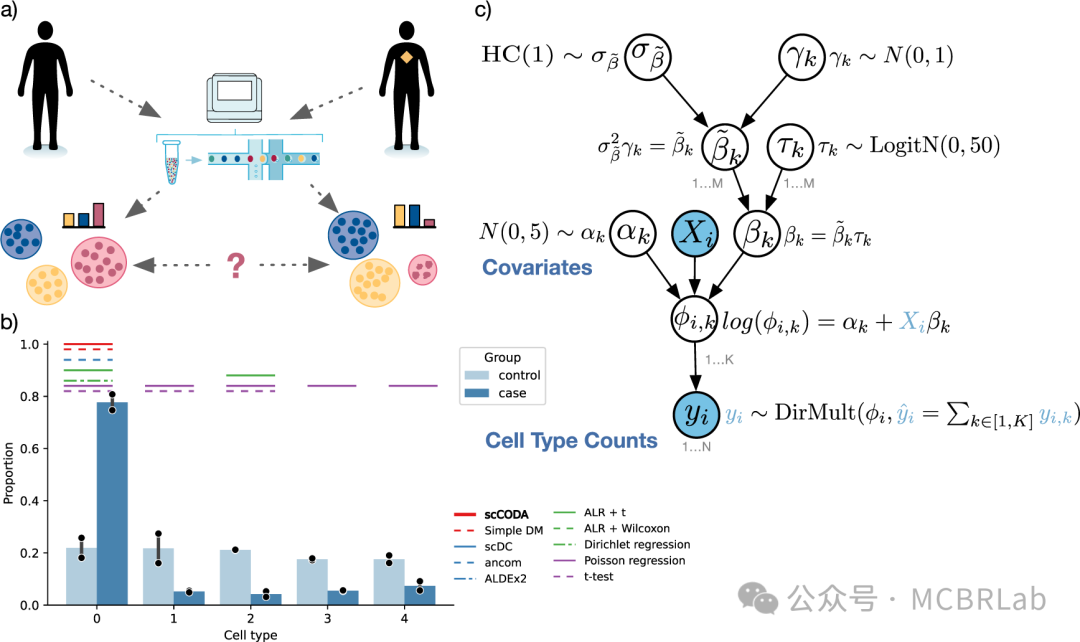
了解疾病病理需要识别和解析受影响的细胞类型。单细胞数据集通过比较疾病细胞与匹配的对照样本以表征疾病引起的细胞变化,为研究疾病机制提供了前所未有的机会。用户利用指标评估细胞类型组成的变化,并结合统计方法获得因扰动而改变的基因和途径,以评估整体分子变化。参考映射方法已成功应用于将来自COVID-19患者支气管肺泡灌洗液(BALF)或人外周血单核细胞(PBMCs)的细胞映射到基于相同或多个组织的健康图谱上,并检测到与疾病相关的细胞类型。此外,来自肾细胞癌(RCC)患者的肿瘤细胞已成功映射到健康肾脏图谱上,揭示了肿瘤区室细胞与参考细胞的分离,而免疫/基质区室则整合到参考中。
如果患病样本显示出在健康参考中不明显的方差,那么稳健地检测疾病中的组成或细胞状态改变可能具有挑战性。因此,将疾病数据集映射到足够大和多样化的健康参考图谱上能够快速识别疾病状态。成功的疾病查询映射应满足以下标准:(1)保留参考中健康细胞状态的异质性;(2)整合参考和查询中的相同细胞类型;(3)保留在疾病数据集中新出现但在参考中不存在的先前未表征的细胞类型和状态。这反映了在映射新样本的背景下,维持生物学变异但减少批次变异的众所周知的权衡。
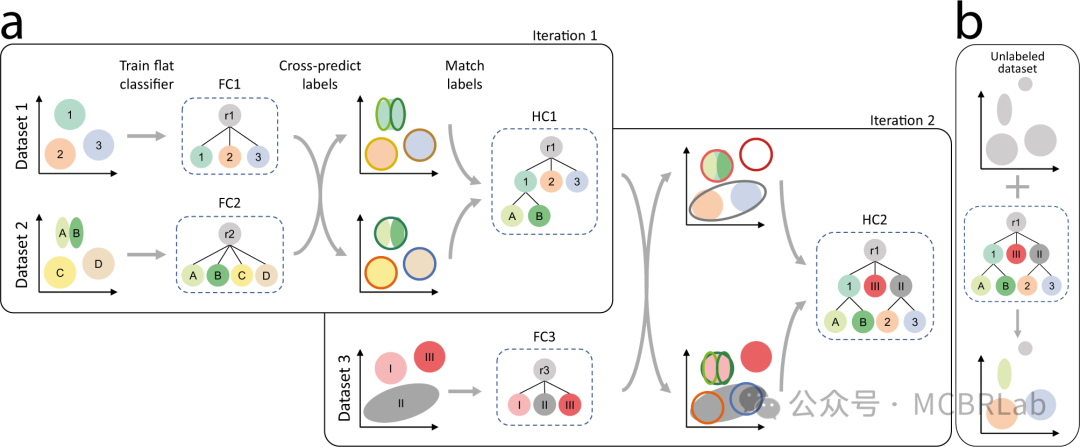
尽管在数据整合方面取得了进展,但疾病状态的自动识别仍然具有挑战性。稳健地恢复定义明确的疾病群体需要一个不确定性度量来区分新的细胞状态与现有参考(图1C)。例如,一个使用距离度量训练的简单K-最近邻(Knn)分类器可以利用从参考到查询的转移标签的不确定性来识别先前未表征的细胞类型和状态。此外,HLCA 作者在SARS-CoV-2肺炎和特发性肺纤维化期间识别出疾病特异性群体。同样,Symphony利用查询细胞与参考细胞的基于细胞或簇的马氏距离来检测未知细胞类型或疾病状态。最近的一种方法将scArches与层次分类器相结合,以学习和扩展细胞类型的层次表示。当添加不适合现有层次结构的查询群体时,其表示能够识别特定群体(如疾病)。这些例子表明,需要新的方法或改进来更稳健地检测疾病状态。使用参考映射进行无监督疾病状态识别与分布外(OOD)检测相关,这是机器学习社区中一个尚未解决的挑战。虽然利用深度生成模型(DGMs)的方法可以基于模型似然来检测 OOD 样本(即查询中的新状态),但它们可能会给 OOD 样本分配比分布内(参考数据)更高的似然。随着这些和其他方法的改进,作者注意到也可以构建特定疾病的参考。虽然构建多个健康和患病参考可能是一项费力的工作,但这种方法将为映射各种查询数据集提供上下文灵活性。
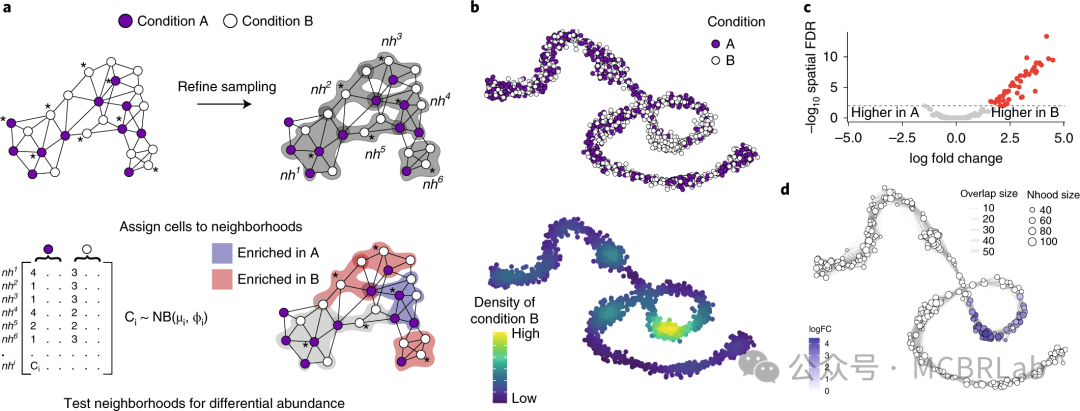
一旦将多个样本映射到共享空间中,开发了一套统计方法来根据对扰动的响应幅度对细胞类型进行优先级排序(即排名)。可以基于扰动数据集中细胞类型比例的变化进行量化,或者基于细胞类型内基因表达变化的幅度进行量化。对于组成变化,MASC和scCODA法基于离散的细胞类型簇识别组成变化,而Milo和MELD使用与细胞类型无关的连续方法在邻域或单细胞水平上量化组成变化。这些方法已被用于分析疾病扰动(如COVID-19、肝硬化或衰老)时发生的组成变化。作为这些工具的补充,还有一些方法专注于通过比较在多种条件下检测到的细胞组的基因表达谱来量化亚群特异性状态转换。例如,Robinson及其同事引入了用于多样本比较的稳健统计工具,而Augur训练分类器可以识别单细胞数据中对扰动最敏感的群体。
总体而言,这些方法展示了scRNA -seq与参考映射相结合如何成为识别复杂群体对扰动如何响应的有力工具。展望未来,作者看到了解决多样本比较可解释性的潜力。疾病尤其不太可能只影响单个细胞亚群,而是会代表在细胞类型间共享且特定于某些细胞状态的复杂响应。能够帮助解析这些差异来源并为下游分析确定特定细胞群体优先级的方法将对更广泛的社区非常有益。
群体规模参考映射
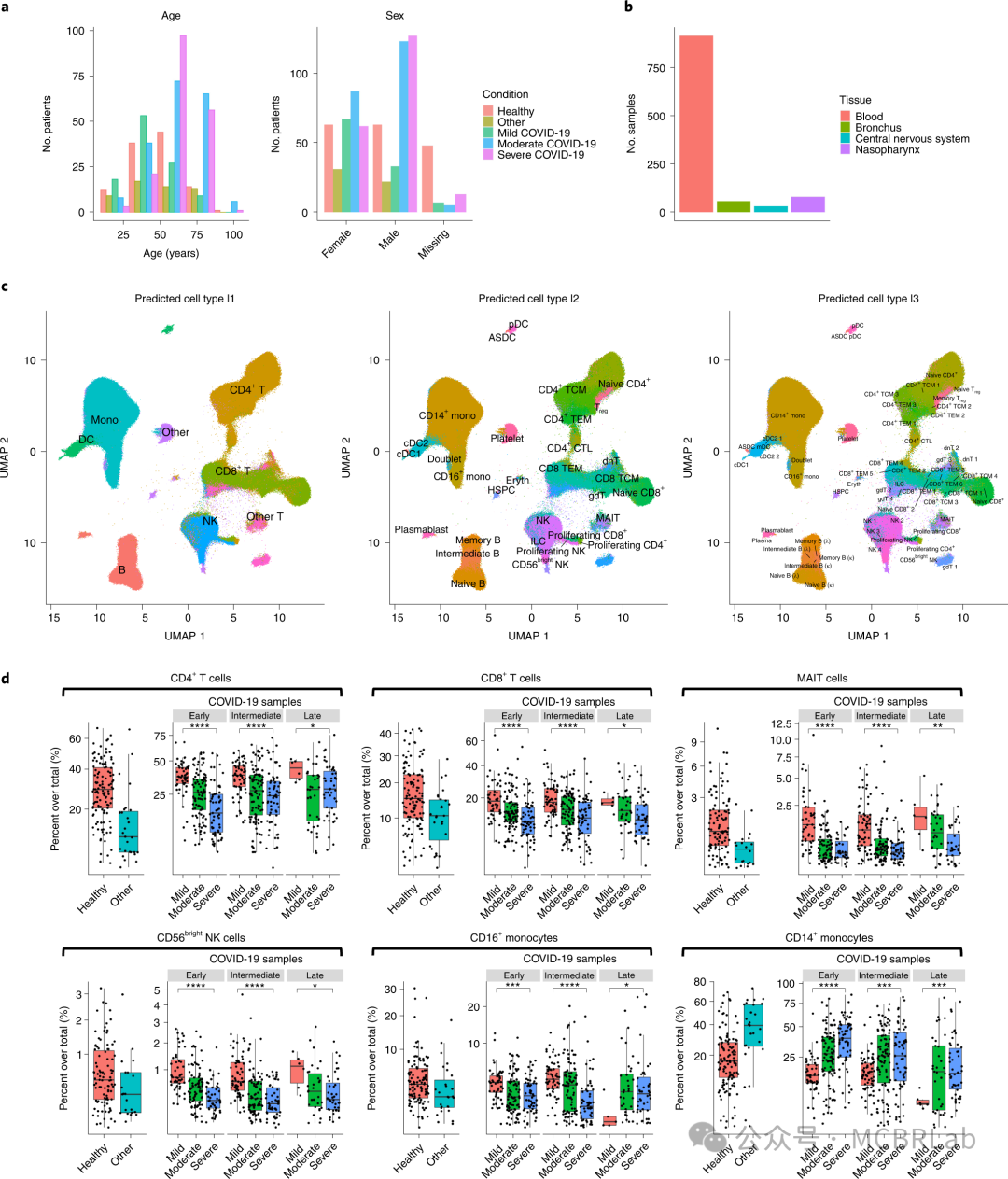
参考映射方法在分析和探索样本群体间大规模变异方面也具有重要潜力。就像人类变异的大型遗传数据库(如gnomAD)在映射到一致的参考框架后对数十万样本进行编目和比较一样,单细胞参考映射工具也能够进行类似的meta分析。一个例子是对 22项COVID-19血液样本的单细胞 RNA 测序研究的meta分析。这些研究总共涵盖了超过 300万个细胞,其年龄、性别、种族、疾病状态和疾病严重程度各不相同。为了便于进行比较,所有样本都被映射到一个单一参考上,从而促进细胞类型标签和元数据的自动协调。标准化有助于进行大规模meta分析,特别是能够识别数百个不同捐赠者中 COVID 诱导的细胞类型组成的可重复变化。由于单细胞研究通常不仅呈现来自大量细胞的数据,还呈现来自大量个体的数据,参考映射可能在解释这些数据集方面发挥关键作用。
除了促进细胞标签的标准化外,参考映射还可能有助于推断和分类查询样本中的疾病状态和严重程度。例如,一类称为多实例学习(MIL)的机器学习算法能够学习这样的映射。MIL算法允许为每个样本(如患者)学习一种转换,并将其作为一个整体进行分类,而无需知道单个标签(受疾病影响或健康)。此外,MIL 方法可以识别导致疾病严重程度的细胞群体。这样的应用将实现疾病严重程度分类的自动化,有助于诊断,同时通过识别每个患者和疾病的疾病相关细胞类型来帮助潜在的(个性化)治疗(图2)。
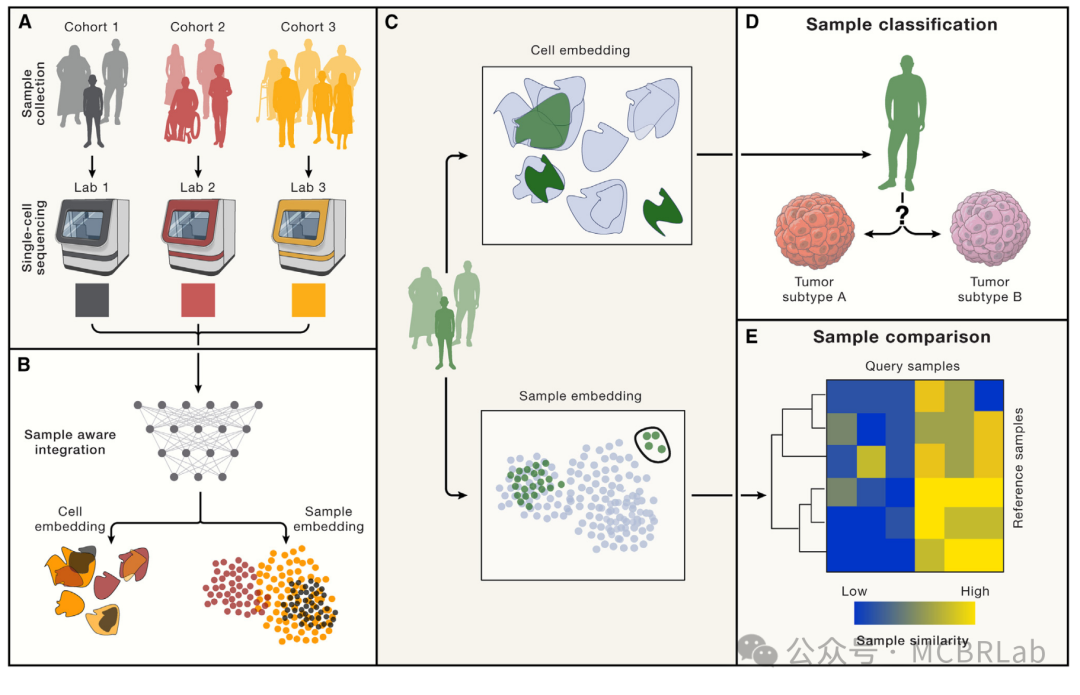
图2. 群体规模的参考映射。(A 和 B) 队列级单细胞参考的可用性使得能够组装由许多样本(或患者)组成的资源,以了解群体和细胞间的异质性(B)。(C) 查询样本被映射到细胞和样本级表示。(D) 在利用细胞嵌入和监督分析映射新样本后,可以对查询样本的疾病表型进行分类(例如,肿瘤类型)。(E) 样本级表示可以推断参考和查询之间直接与细胞级表示相关的样本-样本相似性图。圆圈表示查询中具有不同细胞组成的一组捐赠者,如参考嵌入中所反映的那样。
细胞扰动图谱的构建
单细胞健康图谱越来越多地通过诸如人类细胞图谱等联盟获得。然而,人类细胞图谱生成的数据侧重于健康的稳态条件,而大规模的扰动实验(这里称为 “扰动图谱”)旨在用于药物发现和再生医学,代表了一个新的前沿。将查询数据集映射到扰动图谱的概念在批量研究中已被广泛探索。特别是,大规模批量分子谱分析技术已被用于生成对数千种扰动(包括基因扰动、小分子、细胞因子和药物)的分子响应图谱。Connectivity Map(CMap)、LINCS 1000 和 ChemPert 等数据库将这些扰动组装成数据,并可用于解释广泛的转录特征集。
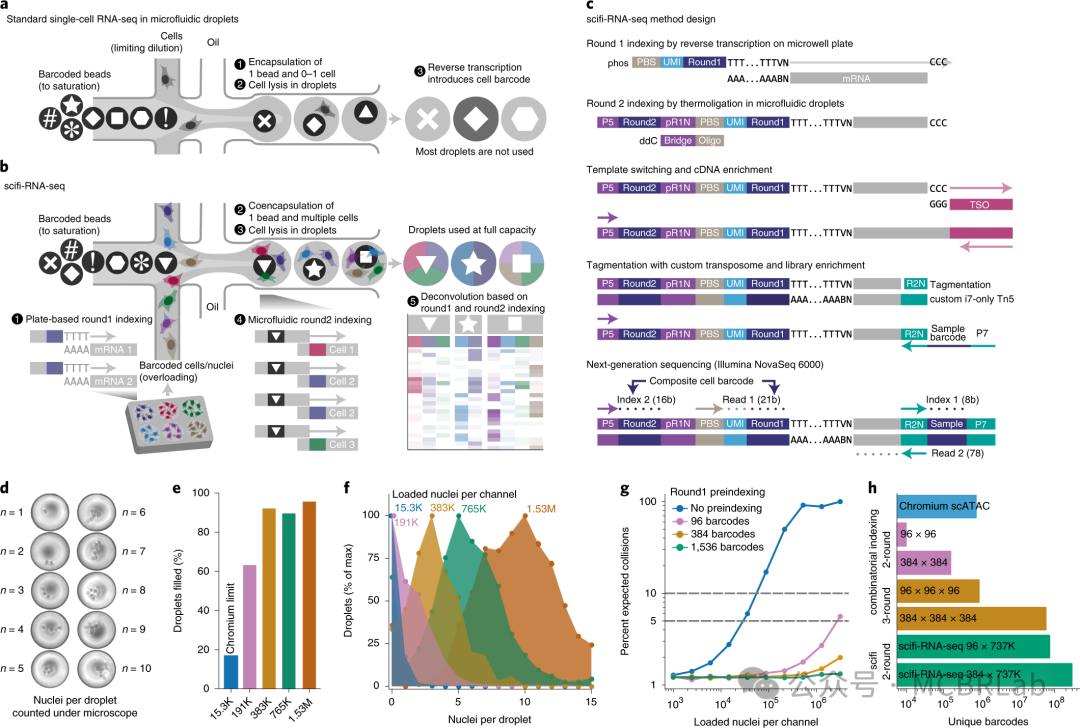
这个概念框架对于以单细胞分辨率测量的扰动具有明显的前景。最近,条形码技术的发展使得能够高通量表征小分子的作用或基于CRISPR-Cas9/13 的单基因或组合基因扰动。这些方法越来越多地应用于类器官系统和诱导多能干细胞(iPSC)衍生模型、体内模型,甚至可以扩展到全基因组扰动实验。随着这些方法的不断发展,参考映射可以帮助将这些数据集与来自健康和患病样本的单细胞图谱联系起来,在实验驱动的扰动和自然观察到的疾病状态之间建立联系。然而,扰动及其组合的探索空间巨大,在实验上无法全部测试(图 1C)。这阻碍了构建与健康图谱类似的全面扰动图谱。数据整合算法可以通过允许将多个稀疏扰动实验整合到一个更全面的图谱中来减少这种情况下的样本稀疏性。虽然整合可能会增加发现能力,但在数据整合和保留生物学变异性之间存在权衡,需要仔细的度量和评估。另一种方法涉及机器学习算法 “插补” 缺失的扰动,使用初始参考数据集来推断先前未见过的扰动对细胞行为的影响。
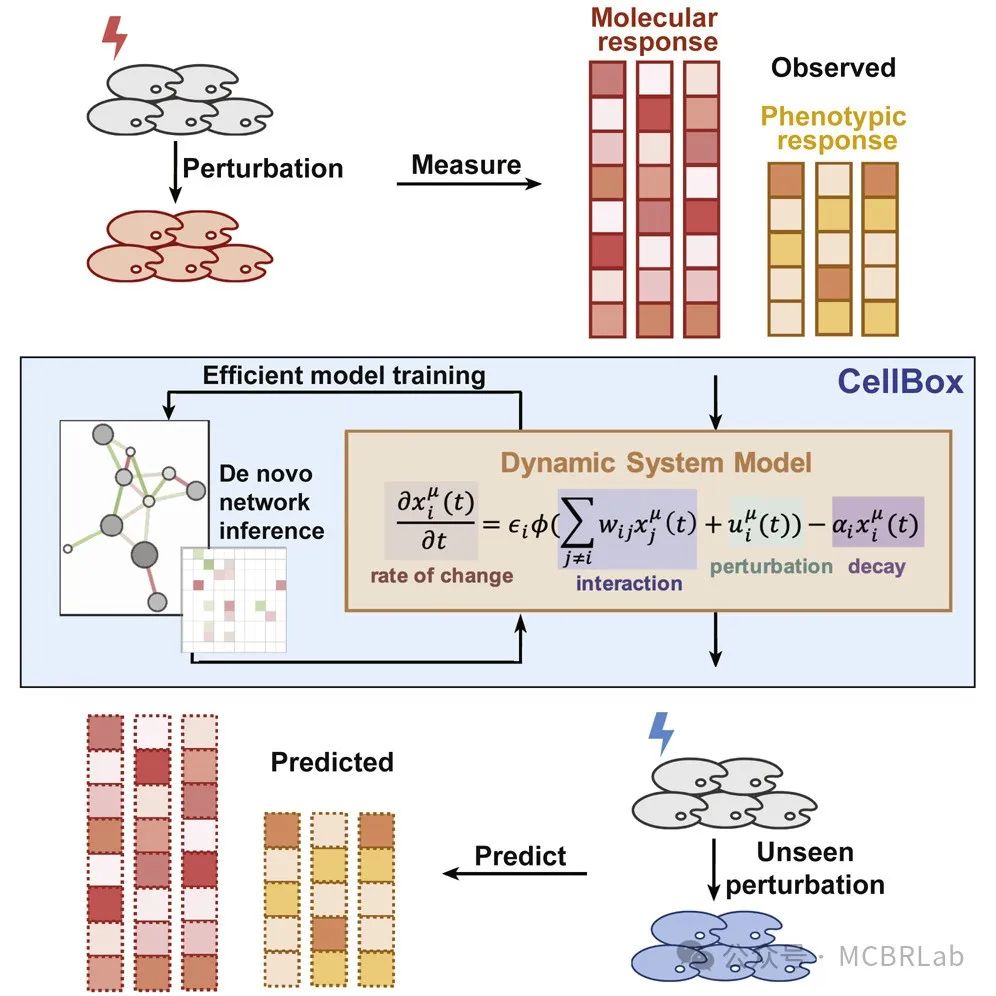
基于动态模型的初步方法已被提出用于预测许多扰动下的增殖测量或基因表达效应。然而,动态方法需要关于调控系统的先验知识来进行模型设计,并且通常依赖于在单细胞水平上难以获得的时间分辨测量。这导致了参数可识别性和拟合挑战。相比之下,线性方法更容易拟合,但对未见过的扰动或复杂的细胞类型特异性行为的泛化能力有限。深度学习方法已被开发来应对这些挑战以预测细胞行为。变分自编码器(VAEs)一直是从单细胞数据中学习低维潜在表示的主要工具。例如,scGen 是一种结合潜在空间向量运算的VAE,用于预测单细胞对不同细胞类型和物种的疾病和化学扰动的响应。在此基础上,组合扰动自编码器(CPA)被提出以扩展现有方法,以预测对药物或基因扰动的组合响应。CPA将细胞表示学习为基础状态与学习到的扰动和协变量(如细胞类型、患者、物种)表示的组合。最后,最近的努力扩展了现有方法以预测先前未分析的化学扰动或基因缺失的影响。所有这些方法都提出了一个清晰的愿景,即预测对单个或组合的未见过的扰动的分子响应,并揭示了扩充扰动图谱的可能途径。未来,将大规模扰动实验与基于深度学习的插补和跨多个研究的整合相结合,可能再加上通过主动学习方法进行的实验扩充,最终实现系统扰动图谱的组装。
跨分子模态的单细胞数据映射
虽然上述技术重点关注将单细胞 RNA 测序(scRNA - seq)查询数据集映射到 scRNA - seq 参考图谱上,但单细胞基因组学领域正迅速转向常规分析其他分子模态。尤其在分析诸如染色质可及性、DNA-蛋白质相互作用图谱或染色体接触相互作用等基因组特征方面存在极大的兴趣。为每种新模态创建一个新的参考数据集以实现查询数据集的映射,这对研究界来说将是一个沉重的负担。因此,人们有兴趣探索跨模态映射的潜力。一个例子是将 scATAC-seq查询数据集映射到由scRNA-seq定义的参考图谱上。如果成功,这些方法将把参考映射框架的广泛优势扩展到scRNA-seq之外的多种模态和技术。跨模态映射的根本挑战在于不同数据集中所测量的特征缺乏对应性。例如,scATAC-seq数据集测量基因组特定区域的染色质可及性,而scRNA-seq 测量基因表达的定量水平。参考数据集和查询数据集之间缺乏重叠特征使得scRNA-seq参考映射工具无法使用,并需要开发新的方法。
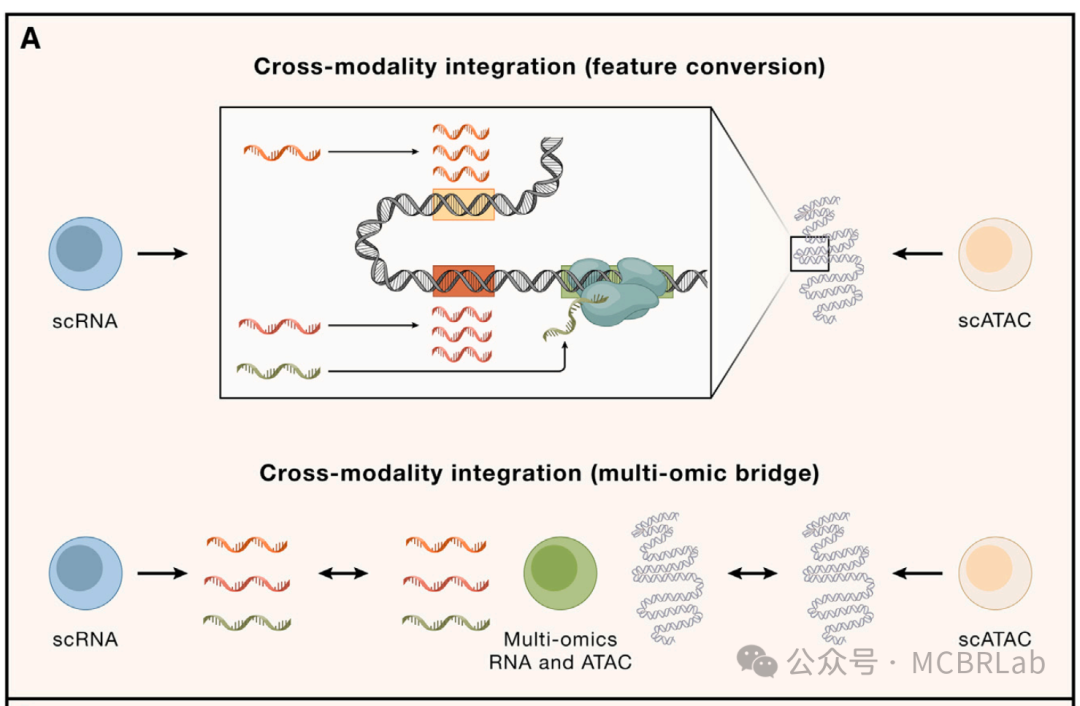
特征转换
第一组跨模态映射方法试图通过将一种测量类型转换为另一种来解决特征对应问题(图 3A)。例如,Cicero算法量化了位于每个基因体及上游2千碱基区域内的ATAC-seq峰的总可及性。注意到位于开放染色质区域的基因往往会积极表达,Cicero 将这些可及性量化值称为“基因活性”分数,它是转录输出的一个替代指标。重要的是,这种特征转换将从ATAC-seq测量的特征转换为scRNA-seq所测量的同一组特征,这代表了向整合迈出的第一步。
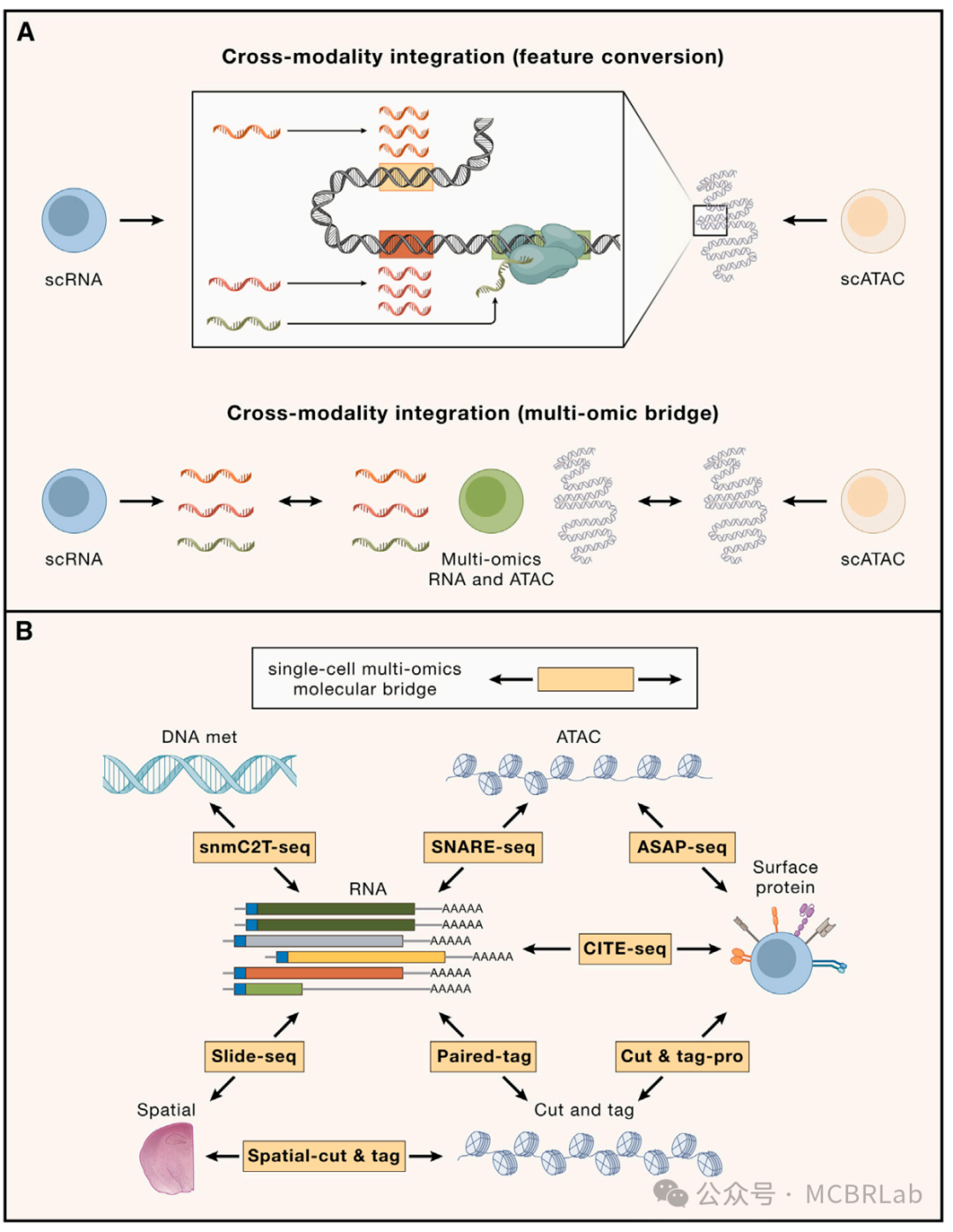
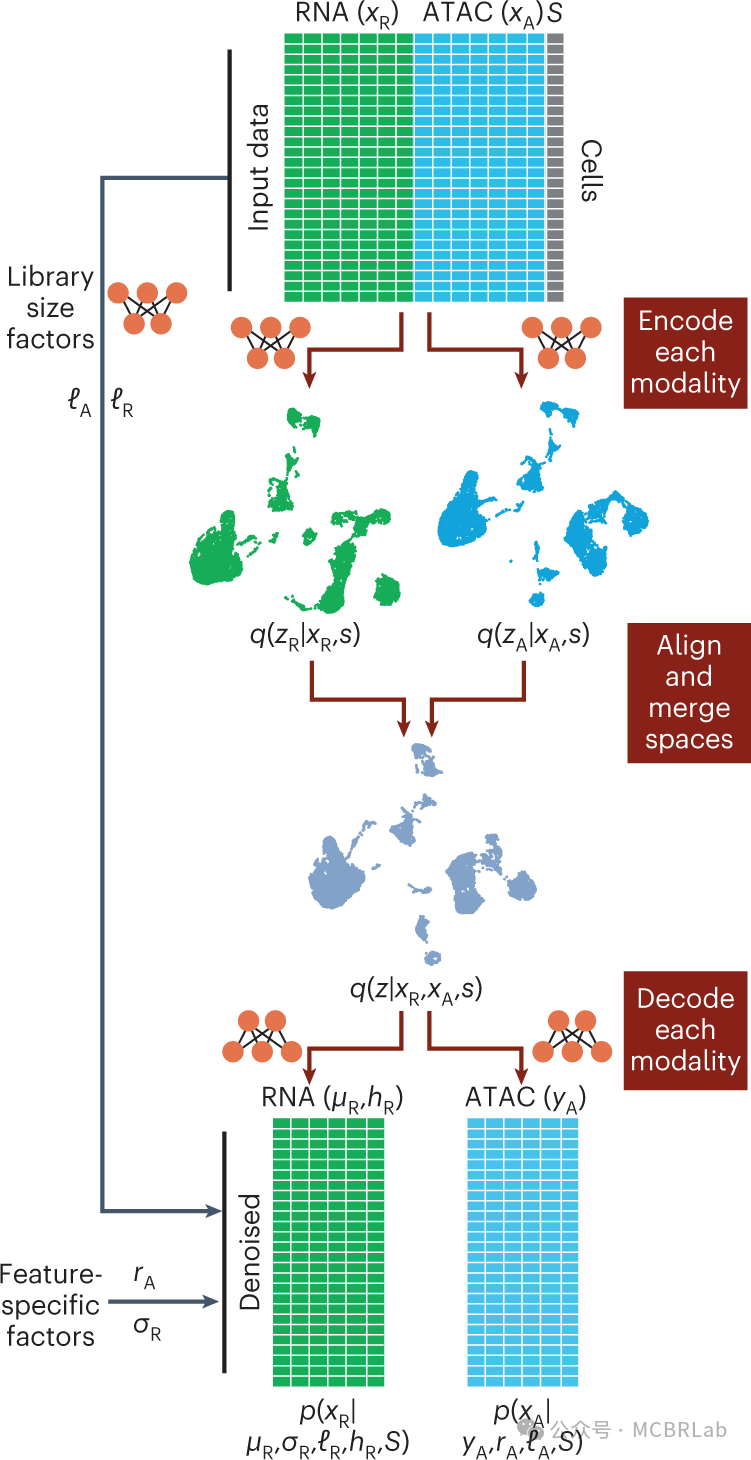
图 3. 跨分子模态的单细胞数据参考映射。(A) 构建跨模态特征对应关系的两种框架。特征转换:将一种测量类型转换为另一种。例如,将基因体内的 ATAC-seq峰转换为基因活性分数,这是 scRNA-seq 测量的相同特征集。多组学桥梁:利用多组学数据集在不同模态之间建立联系。例如,使用同时测量 ATAC 峰和基因表达的数据集连接 ATAC-seq 峰和 RNA-seq 基因。(B) 使用单细胞多组学数据集将RNA参考扩展到其他查询模态。通过使用单细胞多组学技术作为分子桥梁,RNA参考可以扩展到包括其他模态,如DNA甲基化(DNA met)、ATAC 峰、表面蛋白、CUT&Tag(在靶点下切割和标签化)和空间数据。 snmC2T-seq,单核甲基胞嘧啶、染色质可及性和转录组测序;SNARE-seq,单核染色质可及性和 mRNA 表达测序;ASAP - seq,带有选择抗原分析的ATAC测序;CITE-seq,通过测序对转录组和表位进行细胞索引;Paired-Tag,通过测序对单个细胞的 RNA 表达和靶向标签化DNA进行平行分析;CUT&TagPro,带有细胞表面蛋白的单细胞在靶点下切割和标签化;SpatialCUT&Tag,空间在靶点下切割和标签化。
在特征转换之后,现有的整合和映射算法可用于执行跨模态对齐和映射。例如,Seurat v3利用典型相关分析来识别从scATAC-seq和scRNA-seq测量值估计的基因活性分数矩阵之间的保守生物子空间。这个保守子空间能够识别跨数据集的细胞间对应关系,称为锚点。这些锚点使得能够基于已建立的哺乳动物大脑转录组参考图谱对 scATAC-seq图谱进行自动注释。同样,LIGER算法利用非负矩阵分解来推断一组代表跨模态共享生物信号的线性潜在因子。尽管LIGER成功地映射了染色质可及性数据,但DNA甲基化测量值往往与基因表达呈负相关,这也使得能够映射甲基化查询数据集。主要地,Seurat v3和LIGER方法都利用跨模态映射来探索细胞的调控格局与其转录输出之间的关系,从而推断出细胞类型特异性调控网络。
在这些进展的基础上,MultiMAP在特征转换后使用流形学习方法对多个数据集进行降维和整合,推广了UMAP距离度量,以学习一个单一的潜在流形,使来自多种模态的数据均匀分布。另一种方法GLUE实现了一个变分自编码器,用于跨模态的对抗性对齐,由一个基于先验知识的 “引导图” 引导,该图将单个基因组峰与其相关基因连接起来。MultiMAP和GLUE这两种方法都展示了“三重组学”整合的潜力,成功地整合了来自不同细胞的scATAC-seq、scRNA-seq和DNA甲基化图谱。方法的多样性表明了基于特征转换的方法产生有意义映射的潜力。然而,所有这些方法都依赖于转换过程中固有的僵化和简单化的生物学假设。当这些特征相关性假设不成立时,转换方法可能会将不确定性转化为错误。例如,虽然开放染色质通常与活跃转录相关,但情况并非总是如此,特别是在发育系统中,染色质可及性的动态变化和转录输出之间的“滞后” 已有充分记录。
利用多组学数据集进行桥接
跨模态映射的另一种方法是利用最近开发的一系列 “多组学” 单细胞技术,在单个细胞中同时测量一种以上的分子模态。例如,通过测序对转录组和表位进行细胞索引(CITE-seq)利用条形码抗体共同分析单个细胞中的RNA和蛋白质水平,而SHARE-seq、SNARE-seq和10x多组学技术能够对染色质可及性图谱和基因表达水平进行配对的单细胞测量。尽管功能强大,但多组学分析通常比两种单独模态的组合具有更高的财务成本。除此之外,技术噪声的增加和吞吐量的降低限制了其广泛应用。然而,在有多组学图谱的情况下,一系列计算方法可以利用这些数据集来辅助跨模态映射(图 3A)。例如,Seurat v5 通过将多组学数据集用作 “桥梁”来实现跨模态映射。由于桥梁数据集包括在参考数据集和查询数据集中分别表示的模态的配对测量,因此所有参考细胞和查询细胞都可以准确地表示为桥梁细胞的加权组合。这个过程有效地将不同模态的数据集转换为一个共同的特征空间,但不做任何潜在的生物学假设。类似地,StabMap 算法构建了一个连接参考细胞、桥梁细胞和查询细胞的马赛克数据拓扑结构,然后通过识别这个拓扑结构中的最短路径来执行跨模态映射。桥梁整合和StabMap 都展示了多组学桥梁如何与基于特征转换的先前方法相比,大大提高跨模态整合的准确性。此外,它们展示了如何利用不同的桥梁数据集,包括 10X 多组学(scRNA-seq + scATAC-seq)、配对标签(scCUT&Tag+scRNA-seq)和CITE-seq桥梁数据集,能够将各种查询数据集映射到预先存在的 scRNA-seq 参考数据集上。
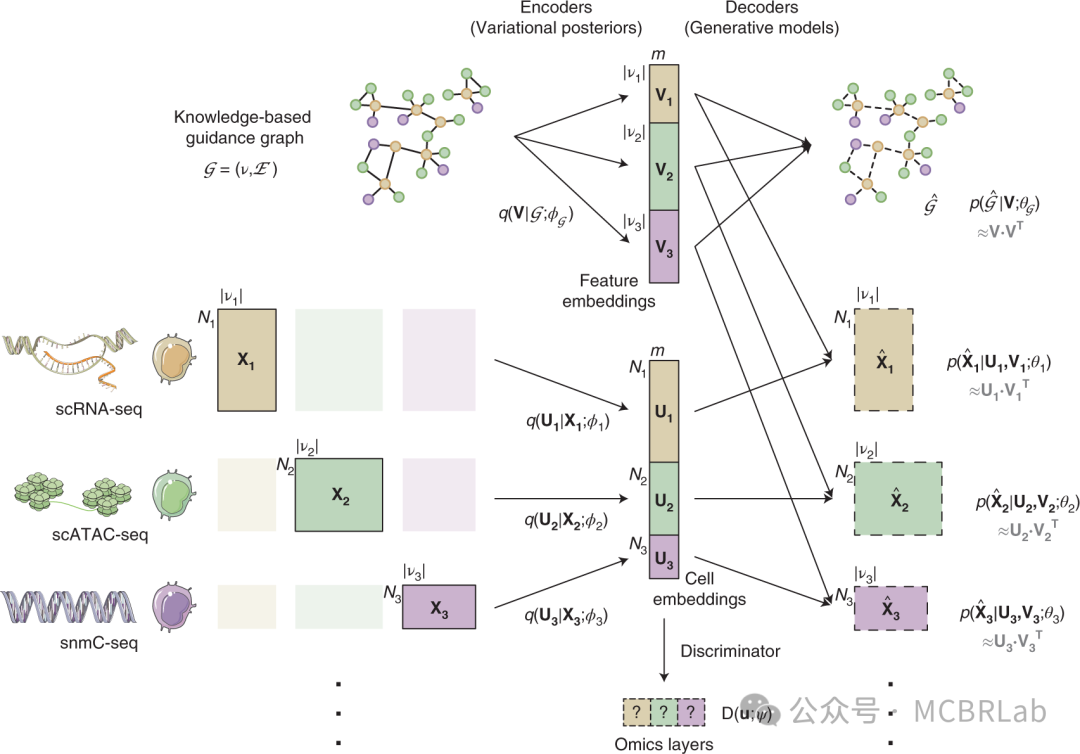
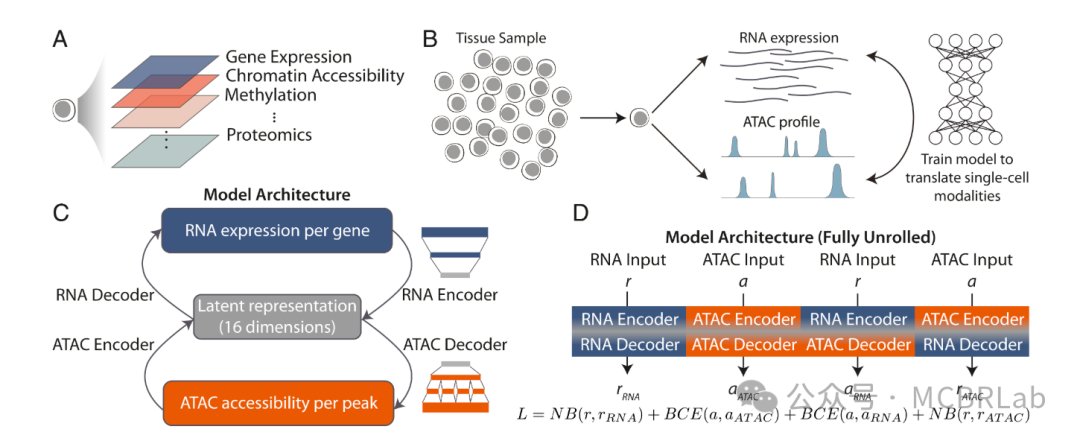
此外,一系列深度学习工具也利用多组学数据集来整合测量不同分子模态的数据集。例如,BABEL 算法利用多组学数据学习一种将一种数据模态映射到另一种数据模态的 “翻译”,基于一个可互操作的神经网络模型。基于这个模型,BABEL可以根据来自另一种模态的测量值为一种模态生成 “预测” 值,并展示了在染色质、RNA 和蛋白质模态之间进行翻译的能力。最近,一系列已发表的作品,如MultiVI、Cobolt 和 CLUE以及Multigrate,都利用多模态变分自编码器(MVAEs)。MVAEs 代表了深度学习的一个最新进展,其中单个神经网络最初对单独的数据集进行单独建模,但随后被投影到一个统一的生物学子空间中。由于这个子空间包含单模态和多模态细胞,这种方法有效地实现了跨模态映射。每种技术都突出了MVAE框架的强大功能,包括定制模态特定的噪声模型(MultiVI)、应用分层生成模型(Cobolt)、应用交叉编码器学习跨模态表示(CLUE)以及在跨模态整合的同时校正批次效应(Multigrate)。
随着跨模态整合工具的不断成熟,作者预计将更加重视用于分析和解释其输出的计算工具。作者也期望进行更系统的比较来评估这些工具,应用关注不同性能指标的各种指标。特别是,跨模态整合实现了一种灵活的实验设计,其中不同的模态在不同的实验中收集,但随后可以一起分析。这可以作为真正的多组学(即同时测量)技术的替代方案,但也可能允许提高每种模态的数据质量和细胞吞吐量。例如,SCENIC + 从染色质可及性和基因表达的配对测量中学习基因调控网络,利用两种模态之间的共变来推断关键转录调节因子及其靶基因。类似地,MultiVelo 整合染色质可及性和基因表达来预测染色质转换和基因剪接状态。这种多模态推断还允许研究人员研究转录因子表达与其结合位点可及性之间的动态关系。虽然最初是为多组学测量开发的,但这些和类似的方法也可以应用于跨模态整合的结果,这将大大拓宽可用于帮助识别跨分子模态关系的数据集的范围。
跨物种映射
单细胞测序现在已经能够在 “全动物” 规模上对整个生物体的细胞进行分子表征,包括蠕虫、果蝇、斑马鱼、青蛙、小鼠,甚至人类胎儿。这些数据集不仅能够对生物体内的细胞异质性进行详细表征,还允许对不同生物体之间的细胞类型和状态进行比较。比较基因组学是注释和识别人类基因组元件的宝贵工具,包括超保守区域和快速进化区域。作者预计单细胞分析将遵循类似的路线图,并进行跨物种分析,以大大提高作者对跨物种共享和独特细胞状态的进化理解。
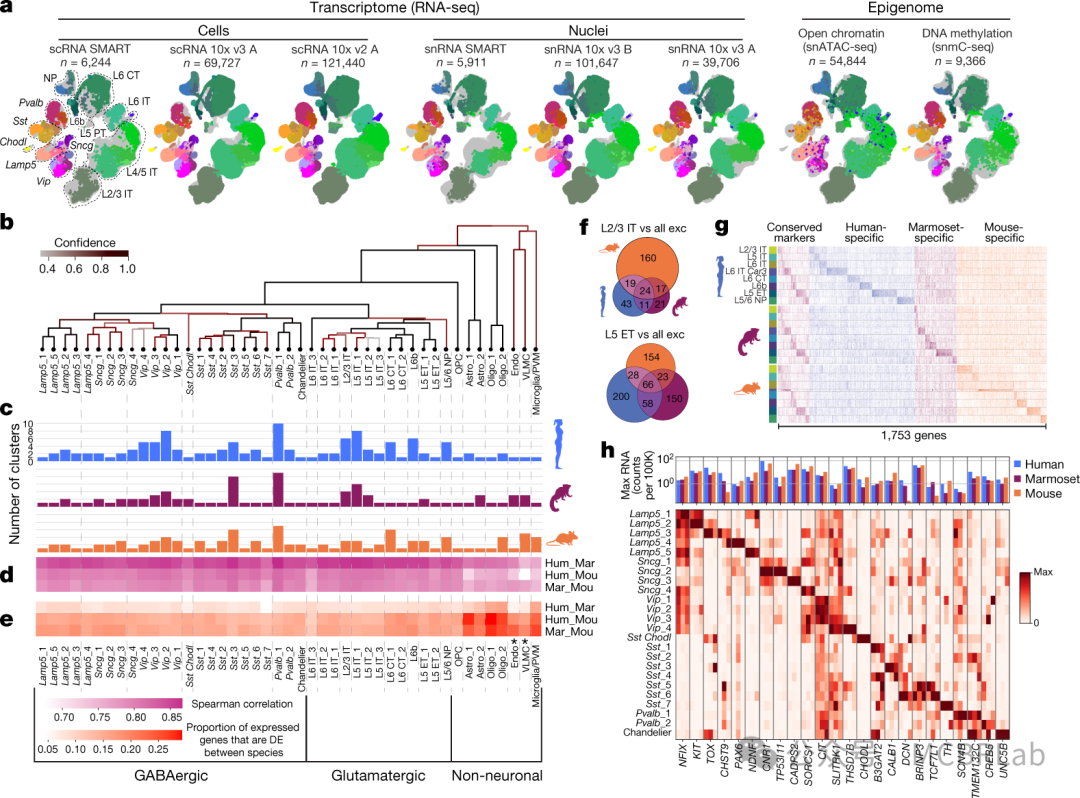
即使存在广泛的转录差异,进化上共享的分子模式也有助于识别跨物种的同源细胞类型。一个这样的例子是在人和小鼠的胰岛中发现了一部分进化上保守的标记物。尽管这些标记物仅占转录组的一小部分,但通过典型相关分析,它们足以准确地对跨物种的细胞类型进行比对。类似的方法已被反复用于探索哺乳动物大脑中细胞类型的进化保守性。例如,跨物种映射工具可用于识别人类和小鼠大脑皮层中兴奋性、抑制性和非神经元细胞类型之间广泛且令人惊讶的保守性。初始比对步骤使得能够详细探究细胞类型丰度和定位的跨物种差异,以及识别大量差异表达的基因模块。这些结果与人类、小鼠和狨猴运动皮层的综合图谱相当。
跨物种比对还能够预测跨物种的细胞特性。一个例子是对梭形神经元(vENs)的解析,它们具有独特的形态,可能与神经精神疾病有关,但其功能特性尚不清楚。通过在人类 scRNA-seq数据中识别出这些细胞的一个罕见群体并进行跨物种映射,哺乳动物初级运动皮层的多模态细胞普查和图谱确定了其与投射到脑下目标的特定的端脑外(ET)兴奋性神经元子集具有很强的同源性。这些发现最终支持了vENs投射到皮质下目标的假设,并指出了vENs可能与之形成回路的特定协作。
跨物种映射方法也有助于识别物种间的明显差异。一个例子是对海龟、蜥蜴和哺乳动物数据集中的神经元 scRNA-seq样本进行比对。令人惊讶的是,它揭示了所有羊膜动物中广泛的γ- 氨基丁酸能中间神经元亚群之间清晰的(一对一)同源性,表明这些细胞类型具有深度的保守性和共同的进化起源。相比之下,在所有物种中都检测到了谷氨酸能神经元,但缺乏明显的分子同源性,这表明其在进化过程中有显著的分化。尽管存在广泛的保守性,但与其他哺乳动物相比,灵长类动物的抑制性中间神经元库存在差异,包括一个丰富的纹状体中间神经元亚群,其与小鼠没有分子同源性。
通常,跨物种映射依赖于一个物种的参考。创建一个通用的多物种参考是增强识别跨物种功能相关和共表达基因的有前景的方法。它甚至可能揭示进化过程中潜在的不同功能。例如,SATURN 算法整合了蛋白质语言模型进化尺度建模2(ESM2),将Aqueous Humor Outflow cell atlas细胞图谱和来自五个物种(人类、食蟹猴、恒河猴、小鼠和猪)的 scRNA - seq数据基于基因表达和蛋白质结构相似性整合到一个共享的低维嵌入空间中。SATURN 的一个显著发现是人类 Myoc 基因的功能与其在其他物种中的同源基因不同。这样的通用参考使作者能够理解众多物种间基因序列和功能的关系。
作者预计跨物种映射方法会持续改进,尽管目前仍具有挑战性,特别是由于缺乏跨物种同源特征的明确定义,以及在广泛的进化变化中识别生物同源性的更广泛挑战。尽管如此,作者期望单细胞分辨率的跨物种分析将继续增进作者对人类细胞类型的功能、独特性和进化起源的理解。特别是,发育过程的跨物种比较为基于分子图谱比较发育阶段提供了有力的机会。此外,替代模态的跨物种比对,特别是通过单细胞 ATAC-seq和单细胞CUT&Tag等技术在单细胞分辨率下测量的染色质特征,将为基因组研究设定新的方向。利用基因组坐标转换将不同物种的基因组坐标从一个物种组装转换到另一个物种组装,这些来自不同物种的染色质特征可以协调到一个统一的基因组空间中,从而实现后续的跨物种比对。功能基因组模态的比对将代表一种独特的方法来注释和表征驱动细胞状态和跨物种分化的调控元件。
基于机器学习的开源图谱构建之路
上文作者概述了参考映射如何实现扰动、多模态、患者队列甚至跨物种数据集的整合。对于该领域而言,一个关键问题是如何制作、发布以及迭代更新参考数据。此外,有多个计划,如人类细胞图谱、人类生物分子图谱计划、生命时间倡议以及陈-扎克伯格倡议等,它们都旨在生成大量数据集并向该领域公开。软件开发中的版本控制,如 Git 和 GitHub,与此类似。为参考模型(例如人类肺细胞图谱 v.1.0.0)分配版本号能够清晰地追踪变化和更新。当生成新数据时,可以发起 “拉取请求”,同时提交参考模型的更新版本(例如 v.1.1.0)以及相应的更新数据。这种机制有助于不同工作组和研究人员对更新内容进行协作评审和整合,确保其准确性和相关性。
此外,随着大规模机器学习模型的涌现以及像Hugging Face这样的开源存储库的发展,这种版本控制方法的实际实施变得可行。研究人员可以利用共享的机器学习模型存储库并为之做出贡献,促进参考模型的协作与民主化。虽然借鉴人类基因组计划来探索基于社区的解决方案颇具吸引力,但基因组参考和单细胞参考之间存在关键差异,这带来了独特的挑战。最为重要的是,人类基因组计划的迭代发布是层层递进的,后续的每个参考版本主要是添加新数据来填补现有空白。这为该领域带来了一个稳定、精心整理且权威的参考基因组。相比之下,单细胞参考的更新通常是对先前版本的细化、修改和补充。这反映了细胞的高度动态性、它们呈现多种状态的能力以及作者对其异质性的不完全理解。
应对这一挑战需要克服目前尚无明确解决方案的后勤和计算难题。例如,多个小组可能最初会发布针对相同人体组织的重叠参考数据集。不同的生物学界也会探索不同的更新参考的方式和时间,比如基于既定时间表或针对新的标志性数据集的产生。与人类基因组计划的标准化不同,这可能会导致针对同一组人体组织出现多种不同的参考,因为它们是由不同的小组生成的。科学界受益于拥有多种测试和迭代改进的选择,过度强制的标准化可能会限制发现的进程。然而,当前的人类细胞图谱参考就像一个缺失许多部分的拼图。与其并行地对相似器官或组织进行分析,开源图谱的理念可以帮助引导实验设计,朝着识别尚未分析且应优先考虑的细胞或组织的方向发展。计算和实验领域的人员作为专注于不同器官的小型网络的一部分共同合作来制定这样的计划至关重要。强制对每个人体组织采用单一的、被社区接受的标准化参考图谱以缓解这一问题是很诱人的。原则上,可以使用基准测试方法来比较不同的参考图谱并选出 “优胜者”。但在实践中,鉴于该领域目前尚处于早期阶段,强制实施严格的标准化可能是有害的。没有一个参考图谱可能是 “完全正确的”,处理相同问题的多个小组可能会生成具有不同或互补优势的参考。生物学界受益于多种测试和迭代改进的选择,过度强制的标准化可能会限制发现的进程。
一种折中的方法可能是有吸引力的,即多个小组可以构建独立的参考图谱,但由选定的机构或领导者监督数据集和资源的最终整合。例如,多个小组已经发布了小鼠大脑的参考图谱,但艾伦脑图谱和美国国立卫生研究院(NIH)的大脑细胞普查网络(BICCN)已牵头将这些小组和数据集整合在一起,以建立一个更全面、更权威的细胞本体。随着这个参考图谱的综合规模不断扩大,应该鼓励(但不强制要求)创建新参考的小组将其数据贡献到这个框架中。然而,确定一个优胜者或将数据集合并到一个参考中并不总是理想的。例如,人类细胞图谱和NIH的LungMAP 倡议都构建了人类肺部的scRNA - seq图谱。这两个计划都汇集了来自广泛多样的实验室和专业知识,用于数据生成、整合和注释。虽然它们生成的图谱有很大的重叠,但它们之间的差异代表了肺部细胞分子状态的前沿发现,如果只选择一个优胜者,这些差异将会减少。单个实验室可以将其数据集映射到这两个图谱上,对比结果并提供反馈,随着时间的推移,这将产生一个更全面、更标准化的参考图谱。
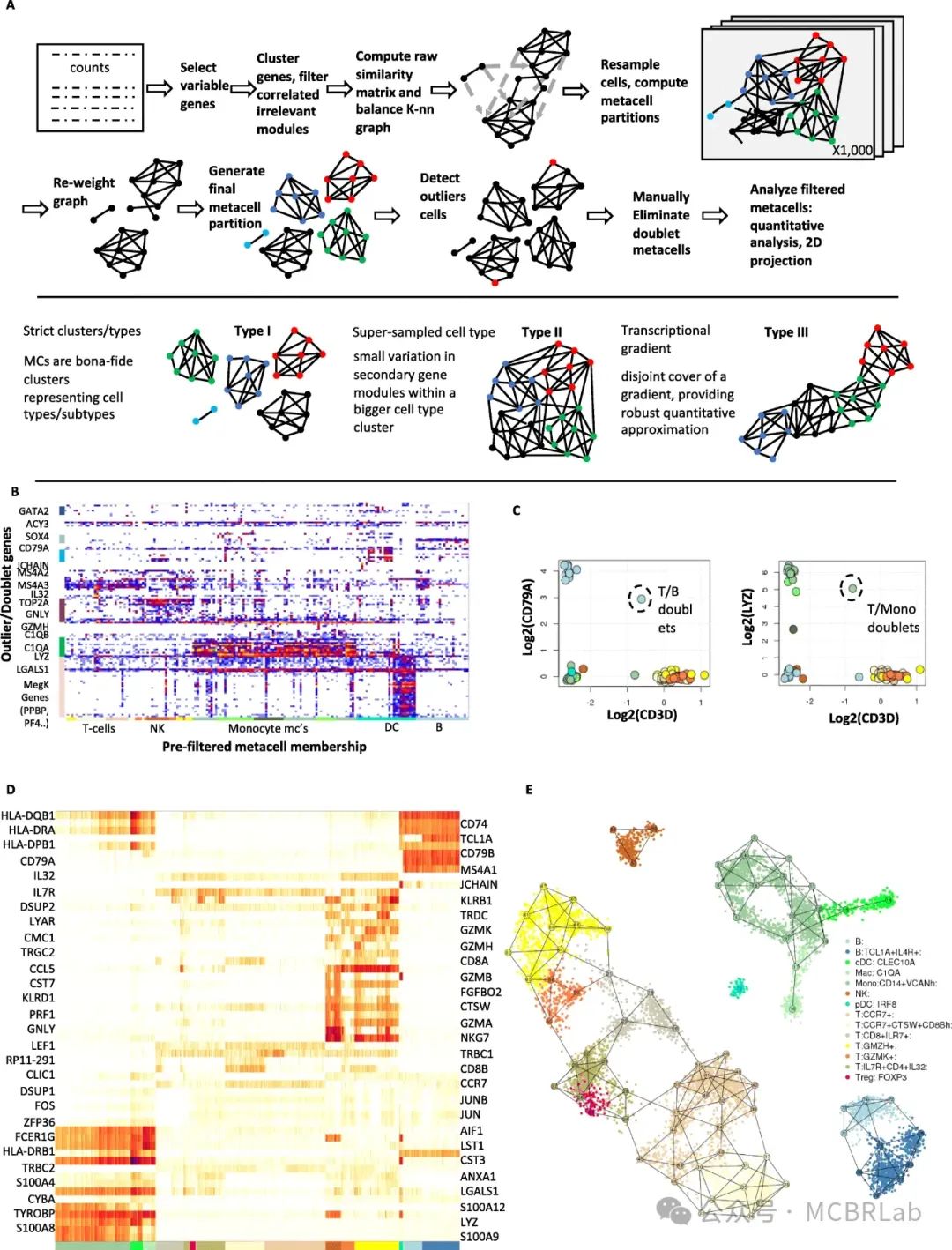
先进的高通量测序技术使得在数百万个细胞中对先前未表征的组织和物种进行详细研究成为可能。然而,对于这些新的生物实体缺乏完善的参考,使得对这些大规模数据集的分析变得复杂。从数百万个细胞构建参考图谱变得至关重要,尤其是当传统方法由于高计算时间和内存需求而失败时。这个挑战可以恰当地称为 “数据压缩”。为此有三个主要策略:(1)将同质细胞聚合成元细胞;(2)从整个数据集中抽取代表性细胞;(3)将整个数据集分割成可管理的块。这些策略旨在在尽量减少计算资源需求的同时保留细胞固有的异质性。值得注意的是,这三种方法并非相互排斥,对于不同的任务可以相互补充。单细胞大语言基础模型等创新进一步拓宽了参考图谱创建的视野,使其在下游分析中的用途更加多样化。
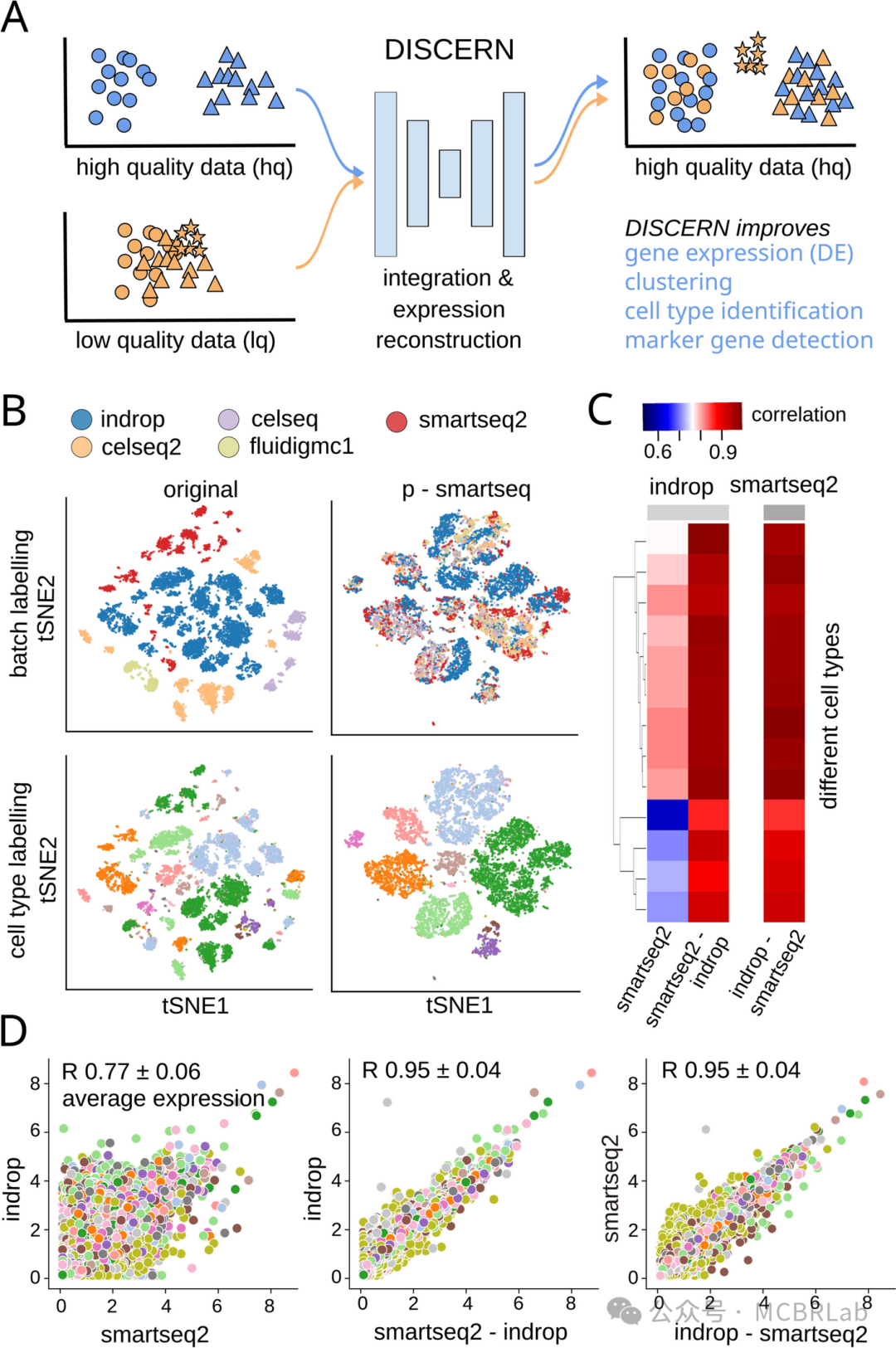
参考映射方法依赖于参考构建算法的质量,这导致了固有的局限性。例如,scArches 依赖于条件生成模型和深度表示学习。这些算法需要大量涵盖各种实验方案的训练数据集,才能有效地对复杂的批次效应进行建模。如果没有足够的数据,它们可能难以映射查询数据集,特别是当查询数据来自参考中未包含的不同技术或物种时。应对这一挑战需要开发在低数据条件下具有良好泛化能力的更稳健的神经网络架构。另一方面,用于参考映射的非深度学习算法(如Seurat和Harmony)可能对数据量需求不大。但它们在处理数千万个数据集时可能会遇到可扩展性问题。克服这一障碍涉及下采样或伪批量处理策略,这可能会在模型中引入偏差。最后,现有的参考映射算法主要在潜在空间中运行,而不是在经过校正的特征矩阵中。为了能够使用校正后的特征矩阵进行下游分析,必须开发能够直接在输入空间中运行的更稳健的参考构建算法。
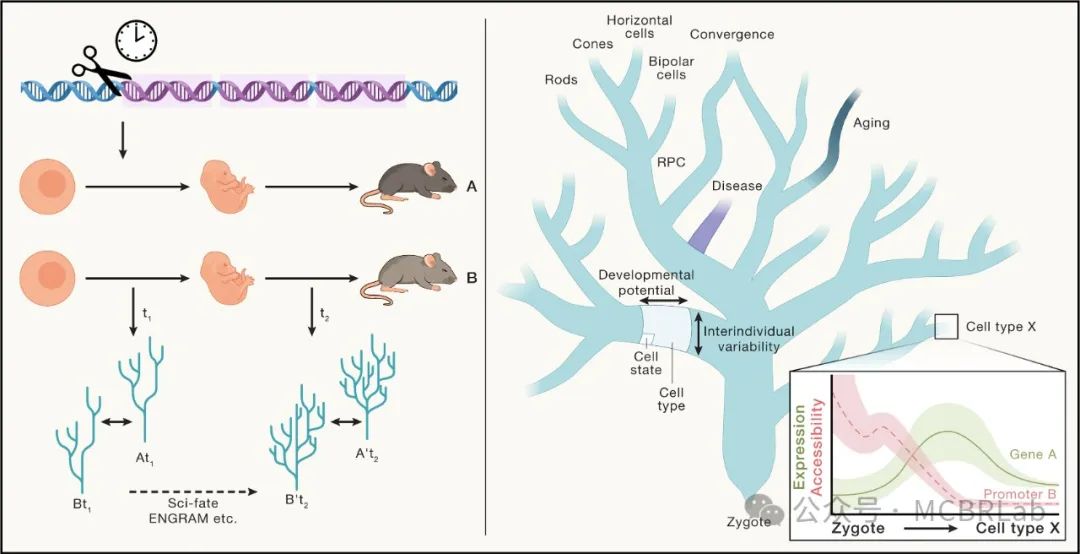
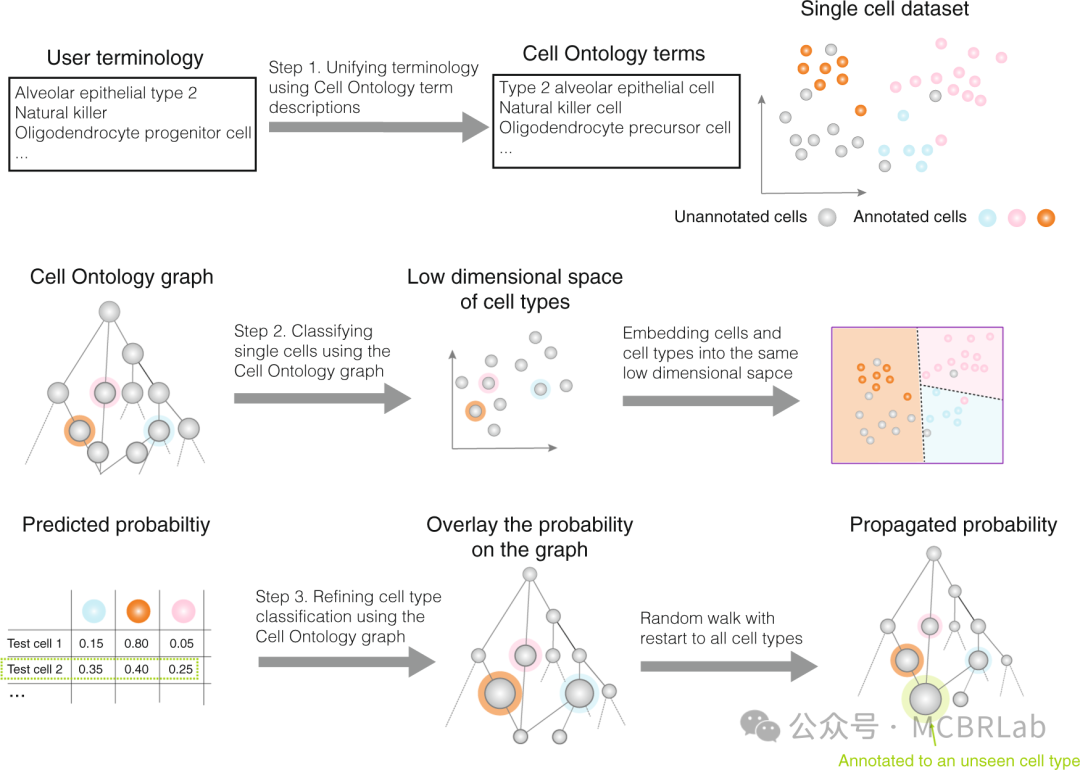
除了计算方面的障碍外,从参考数据向查询数据转移知识的有效性还受到参考元数据质量的影响,特别是细胞类型注释。在一个器官有多个参考数据,且每个参考数据由不同小组用不同的注释集进行标注的情况下,这一点尤为重要。注释的多样性使得选择最合适的图谱成为一项挑战。因此,采用更系统的方法对于在类似参考数据之间建立共识注释至关重要。参考细胞本体或类似于 “参考细胞树”的框架有助于协调和整合不同的注释来源,使其成为一个连贯的集合(树)。这种整合缓解了细胞类型和状态的临时命名问题。可以采用机器学习方法来构建并持续更新这些层次化参考数据,为每个器官分配一棵树。这种有原则且统一的方法使从业者能够系统地命名和注释细胞类型及状态。
文中所描述的示例和用例凸显了参考映射算法在改变用户分析、解释和探索单细胞数据的基本分析流程方面的巨大潜力。展望未来,作者预计参考映射将缓慢但肯定地开始取代无监督聚类和手动注释工作流程。如此一来,单细胞分析将从以专家为中心且繁琐的流程转变为对初学者和专家而言都快速、易用且准确的程序
如何学习大模型 AI ?
由于新岗位的生产效率,要优于被取代岗位的生产效率,所以实际上整个社会的生产效率是提升的。
但是具体到个人,只能说是:
“最先掌握AI的人,将会比较晚掌握AI的人有竞争优势”。
这句话,放在计算机、互联网、移动互联网的开局时期,都是一样的道理。
我在一线互联网企业工作十余年里,指导过不少同行后辈。帮助很多人得到了学习和成长。
我意识到有很多经验和知识值得分享给大家,也可以通过我们的能力和经验解答大家在人工智能学习中的很多困惑,所以在工作繁忙的情况下还是坚持各种整理和分享。但苦于知识传播途径有限,很多互联网行业朋友无法获得正确的资料得到学习提升,故此将并将重要的AI大模型资料包括AI大模型入门学习思维导图、精品AI大模型学习书籍手册、视频教程、实战学习等录播视频免费分享出来。

第一阶段(10天):初阶应用
该阶段让大家对大模型 AI有一个最前沿的认识,对大模型 AI 的理解超过 95% 的人,可以在相关讨论时发表高级、不跟风、又接地气的见解,别人只会和 AI 聊天,而你能调教 AI,并能用代码将大模型和业务衔接。
- 大模型 AI 能干什么?
- 大模型是怎样获得「智能」的?
- 用好 AI 的核心心法
- 大模型应用业务架构
- 大模型应用技术架构
- 代码示例:向 GPT-3.5 灌入新知识
- 提示工程的意义和核心思想
- Prompt 典型构成
- 指令调优方法论
- 思维链和思维树
- Prompt 攻击和防范
- …
第二阶段(30天):高阶应用
该阶段我们正式进入大模型 AI 进阶实战学习,学会构造私有知识库,扩展 AI 的能力。快速开发一个完整的基于 agent 对话机器人。掌握功能最强的大模型开发框架,抓住最新的技术进展,适合 Python 和 JavaScript 程序员。
- 为什么要做 RAG
- 搭建一个简单的 ChatPDF
- 检索的基础概念
- 什么是向量表示(Embeddings)
- 向量数据库与向量检索
- 基于向量检索的 RAG
- 搭建 RAG 系统的扩展知识
- 混合检索与 RAG-Fusion 简介
- 向量模型本地部署
- …
第三阶段(30天):模型训练
恭喜你,如果学到这里,你基本可以找到一份大模型 AI相关的工作,自己也能训练 GPT 了!通过微调,训练自己的垂直大模型,能独立训练开源多模态大模型,掌握更多技术方案。
到此为止,大概2个月的时间。你已经成为了一名“AI小子”。那么你还想往下探索吗?
- 为什么要做 RAG
- 什么是模型
- 什么是模型训练
- 求解器 & 损失函数简介
- 小实验2:手写一个简单的神经网络并训练它
- 什么是训练/预训练/微调/轻量化微调
- Transformer结构简介
- 轻量化微调
- 实验数据集的构建
- …
第四阶段(20天):商业闭环
对全球大模型从性能、吞吐量、成本等方面有一定的认知,可以在云端和本地等多种环境下部署大模型,找到适合自己的项目/创业方向,做一名被 AI 武装的产品经理。
- 硬件选型
- 带你了解全球大模型
- 使用国产大模型服务
- 搭建 OpenAI 代理
- 热身:基于阿里云 PAI 部署 Stable Diffusion
- 在本地计算机运行大模型
- 大模型的私有化部署
- 基于 vLLM 部署大模型
- 案例:如何优雅地在阿里云私有部署开源大模型
- 部署一套开源 LLM 项目
- 内容安全
- 互联网信息服务算法备案
- …
学习是一个过程,只要学习就会有挑战。天道酬勤,你越努力,就会成为越优秀的自己。
如果你能在15天内完成所有的任务,那你堪称天才。然而,如果你能完成 60-70% 的内容,你就已经开始具备成为一名大模型 AI 的正确特征了。
这份完整版的大模型 AI 学习资料已经上传CSDN,朋友们如果需要可以微信扫描下方CSDN官方认证二维码免费领取【保证100%免费】


















 1764
1764

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









