






hello,大家好,今天我们来分享多组学数据推断RNA Velocyto,大家做velocyto的时候估计都是用RNA数据来分析的,其实,对应的ATAC数据也含有丰富的动力学信息,多组学推断更加准确一点,参考的文献在Single-cell multi-omic velocity infers dynamic and decoupled gene regulation,非常棒的文章,我们先来分享文章,最后看看示例代码。
Abstract
单细胞多组学数据集,其中在同一细胞内分析多种分子模式,提供了一个独特的机会来发现细胞表观基因组和转录组变化之间的关系(这里我们就是指 RNA + ATAC)。为了实现这一目标,开发了 Mu
ltiVelo,这是一种基因表达机制模型,可扩展 RNA 速度框架以纳入表观基因组数据。 MultiVelo 使用概率潜在变量模型从单细胞数据中估计染色质可及性和基因表达的转换时间和速率参数,提供表观基因组和转录组变化之间时间关系的定量总结。与仅从 RNA 估计的速度相比,结合染色质可及性数据显着提高了细胞命运预测的准确性。在来自大脑、皮肤和血细胞的单细胞多组学数据集上拟合 MultiVelo 揭示了两类不同的基因,其区别在于染色质是在转录停止之前还是之后关闭。MultiVelo模型还识别了四种类型的细胞状态——两种表观基因组和转录组耦合的状态和两种不同的解耦状态。 MultiVelo 推断的参数量化了基因占据四种状态中每一种状态的时间长度,通过转录组和表观基因组之间的耦合程度对基因进行排序。最后,分析确定了转录因子表达和结合位点可及性之间以及疾病相关 SNP 可及性和相关基因表达之间的时间滞后。
Introduction
从 DNA 到 RNA 再到蛋白质的基因表达调控是控制细胞命运的关键过程。协调的、逐步的基因表达变化——基因按特定顺序打开和关闭——是细胞特化发育过程的基础。 越来越多的高通量单细胞测序技术被用于揭示这些逐步的基因表达变化。 但是,由于实验测量会破坏细胞,因此只能进行时间快照测量,并且不可能观察到相同的单个细胞随时间的变化。
计算方法可以利用单细胞快照来推断发育过程中的顺序基因表达变化。例如,细胞轨迹推断算法使用成对的细胞相似性将细胞映射到与预测的发育进展相对应的“伪时间”轴上。然而,基于相似性的轨迹推断无法预测细胞转变的方向或相对速率。推断 RNA Velocyto的方法通过拟合微分方程系统来解决这些限制,该系统使用剪接和未剪接的转录物计数来描述转录变化的方向和速率。最初的 RNA Velocyto方法依赖于稳态假设来拟合模型参数,但后来的工作开发了一个动力学模型(就是我们常用的Scvelo) ,除了稳态外,该模型明确适合基因表达的诱导和抑制阶段。至关重要的是,这种 RNA Velocyto的动力学模型还推断出每个细胞的潜在时间值,提供了一种在细胞分化过程中重建基因表达变化顺序的机械手段。最近的一篇论文进一步扩展了 RNA 速度框架,以包括来自相同细胞的基因表达和蛋白质测量,但使用稳态假设来估计参数,因此没有估计每个细胞的潜伏时间值。单细胞表观基因组值也被单独用于推断细胞分化的未来方向,但这些方法没有结合基因表达。
单细胞多组学测量提供了将表观基因组数据纳入转录机制模型的机会。 例如,SNARE-seq、SHARE-seq 和 10X Genomics Multiome 等新技术可以量化同一细胞中 RNA 和染色质的可及性。 表观基因组和转录组在细胞分化过程中都会发生变化,因此单细胞多组学数据集中的时间快照可能揭示这些分子层之间的相互作用。 例如,如果表观基因组谱系启动发生在特定的基因组位点,单细胞多组学数据可能会揭示基因的染色质重塑与其转录之间存在显着的时间滞后。 同样,观察转录因子表达和假定结合位点染色质可及性的动态变化可以揭示它们的时间关系。
现有的 RNA 速度模型假设基因的转录率在基因表达的整个诱导阶段是一致的。 然而,表观基因组的变化在调节基因表达方面起着关键作用,例如收紧或放松启动子和增强子区域的染色质compaction。 例如,从常染色质到异染色质的转变会显着降低该位点的转录率,因为转录机制无法访问 DNA。 因此,一个更现实的模型将反映增强子和启动子染色质可及性对转录率的影响 。
开发了 MultiVelo,这是一种计算方法,用于从单细胞多组学数据集中推断基因表达的表观基因组调控。 模型扩展了动态 RNA 速度模型以结合多组学测量,以更准确地预测每个细胞的过去和未来状态,共同推断每种模式的瞬时诱导或抑制率,并确定模式之间的耦合程度或时间滞后 。MultiVelo 使用概率潜在变量模型来估计基因调控的转换时间和速率参数,提供表观基因组和转录组变化之间时间关系的定量总结。
示例证明 MultiVelo 准确地恢复了细胞谱系,并量化了染色质可及性和基因表达暂时不同步的引发和去耦间隔的长度。 微分方程模型准确地拟合了来自胚胎小鼠大脑、胚胎人类大脑的单细胞多组学数据集,以及来自人类造血干细胞和祖细胞的新生成的数据集。 此外,模型通过染色质可及性预测了两种不同的基因表达调控机制,并且在研究的所有组织中确定了两种机制的明确例子。 最后,使用 MultiVelo 来推断转录因子 (TF) 与其结合位点之间以及 GWAS SNP 与其连锁基因之间的时间关系。 总之,MultiVelo 提供了对表观基因组变化在细胞命运转变过程中调节基因表达的机制的基本见解。
Results
MultiVelo: A Mechanistic Model of Gene Expression Incorporating Chromatin Accessibility(这部分内容涉及到很多数学符号,就不给大家翻译了)


也就是说估计三部分,ATAC,pre-RNA,RNA。
MultiVelo 模型的数学公式立即导致了关于基因表达过程中染色质可及性与转录之间关系的两个重要见解。 首先,染色质可及性和 RNA 转录状态有多种数学上可行的组合。 也就是说,当转录被诱导或抑制时,染色质可以打开或关闭。 这意味着多个事件顺序是可能的:染色质关闭可以在转录抑制开始之前或之后发生。 将第一个排序(染色质关闭在转录抑制之前开始)称为模型 1,将第二个排序称为模型 2。Note that there are other mathematically possible orderings where transcription occurs before chromatin opening, but these are not biologically plausible, and we do not find convincing evidence that they occur in the datasets we analyzed。

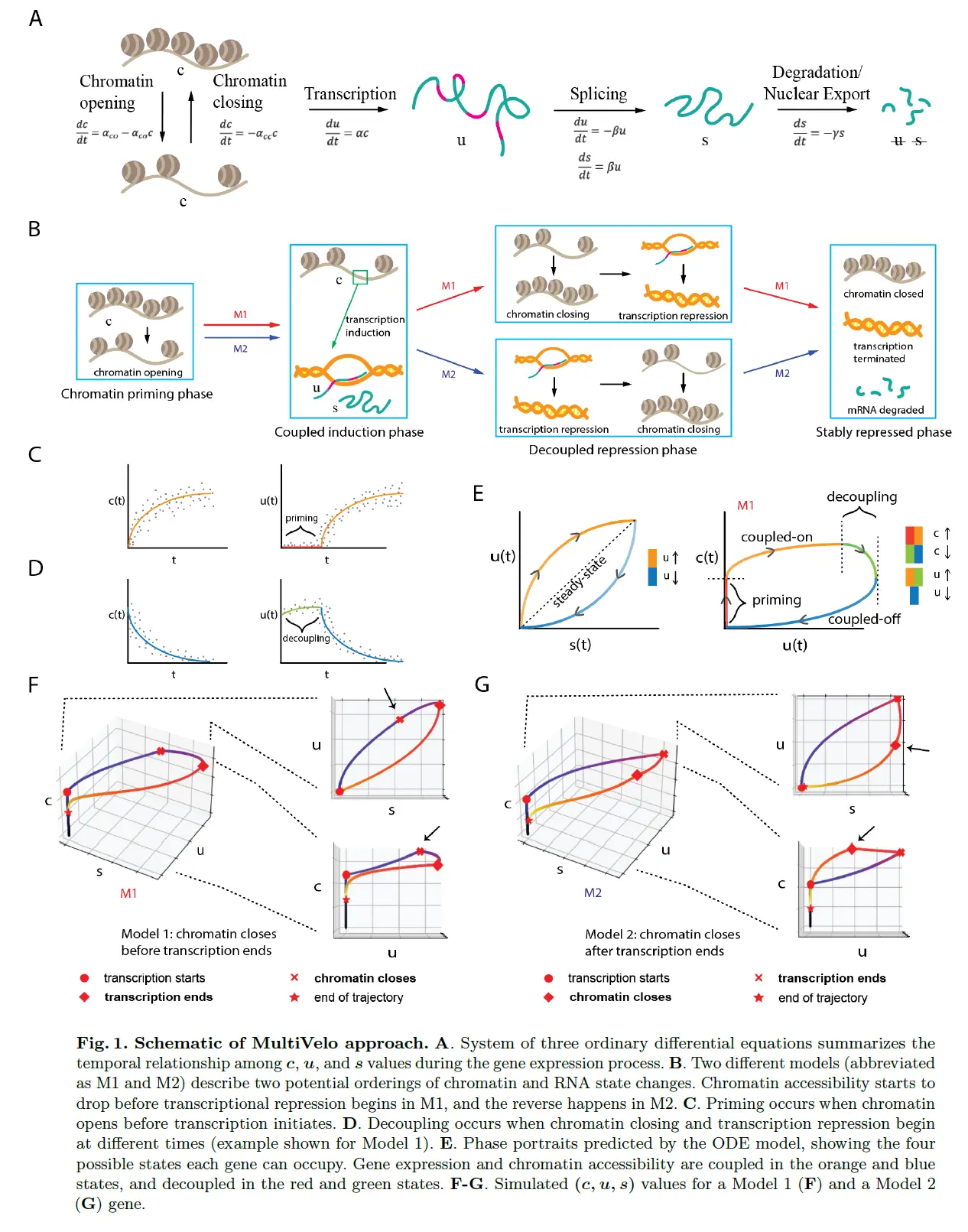
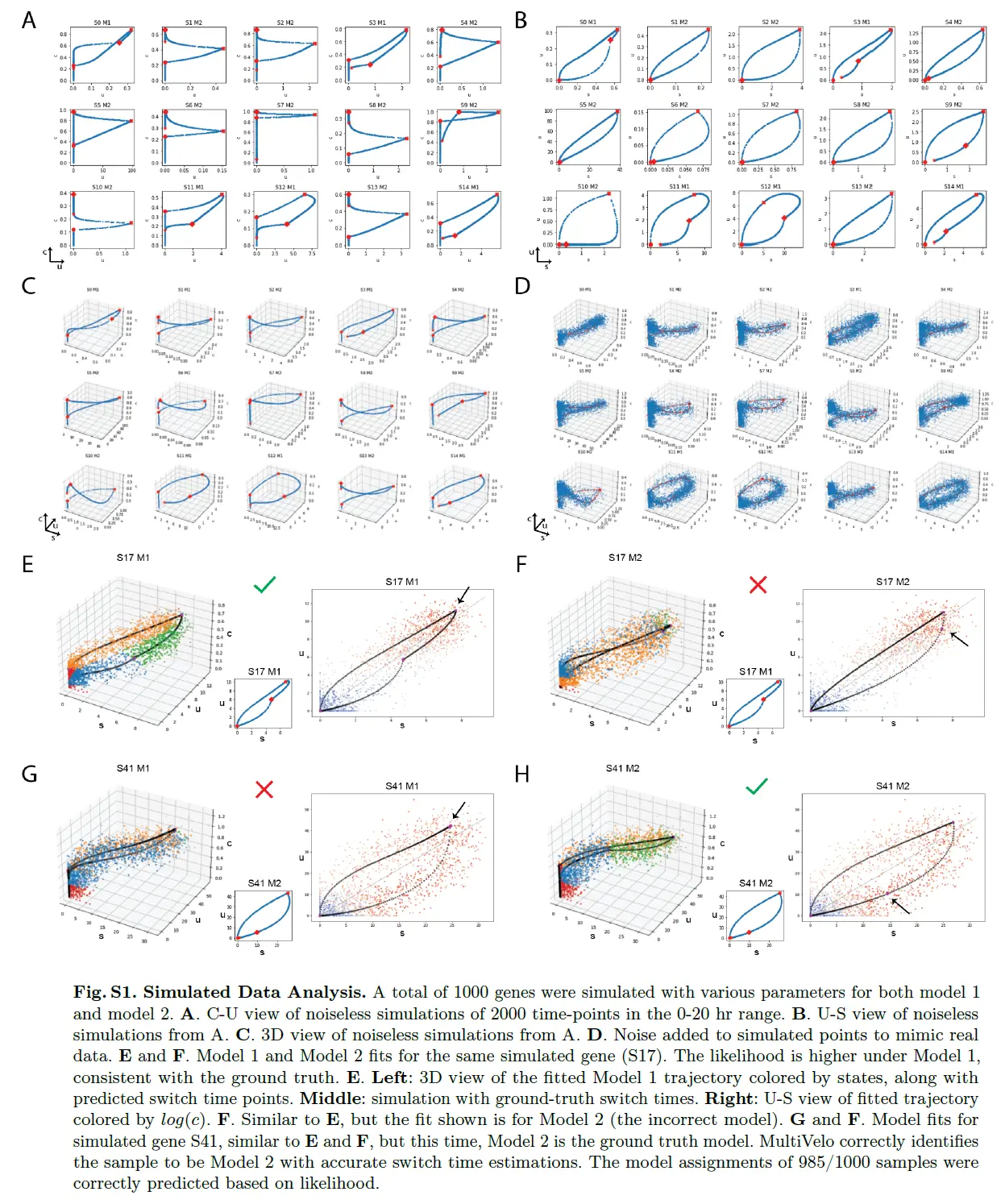
MultiVelo Accurately Fits Simulated Data
进行了模拟,以确定 MultiVelo 是否可以恢复速率参数和切换时间,并在存在噪声的情况下区分模型 1 和模型 2。 结果表明,MultiVelo 可以准确拟合噪声数据,并且可以恢复底层参数。 此外,发现 MultiVelo 以高精度区分模型 1 和模型 2(98.5% 的模拟基因根据模型似然被正确分配)。 分析还证实,在拟合 ODE 参数之前,可以通过简单地比较稳态线上方和下方顶部分位数中的细胞数量来区分模型 1 和模型 2 基因(95.8% 的模拟基因被正确分配 ) 。
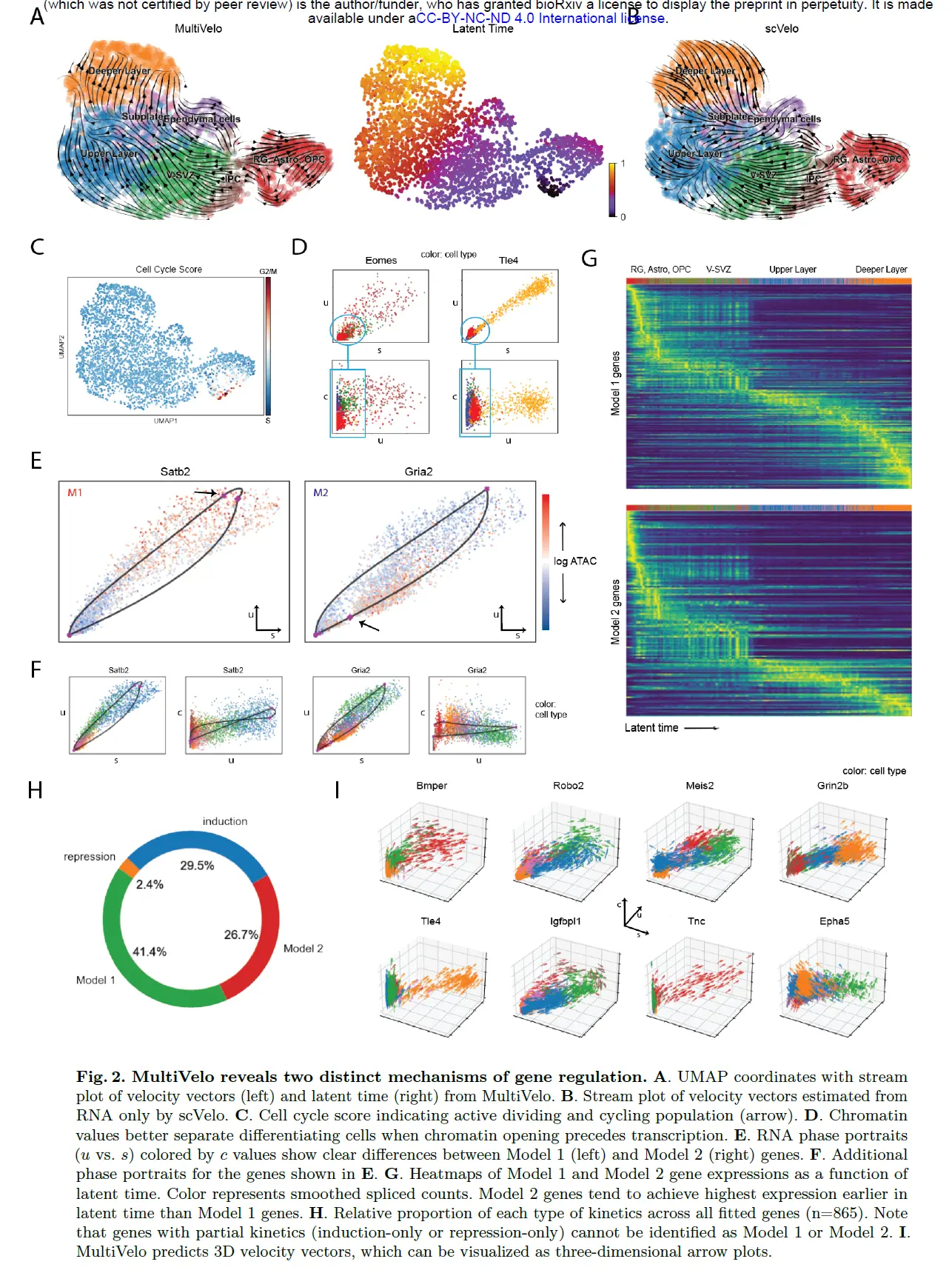
MultiVelo Distinguishes Two Models of Gene Expression Regulation in Embryonic Mouse Brain
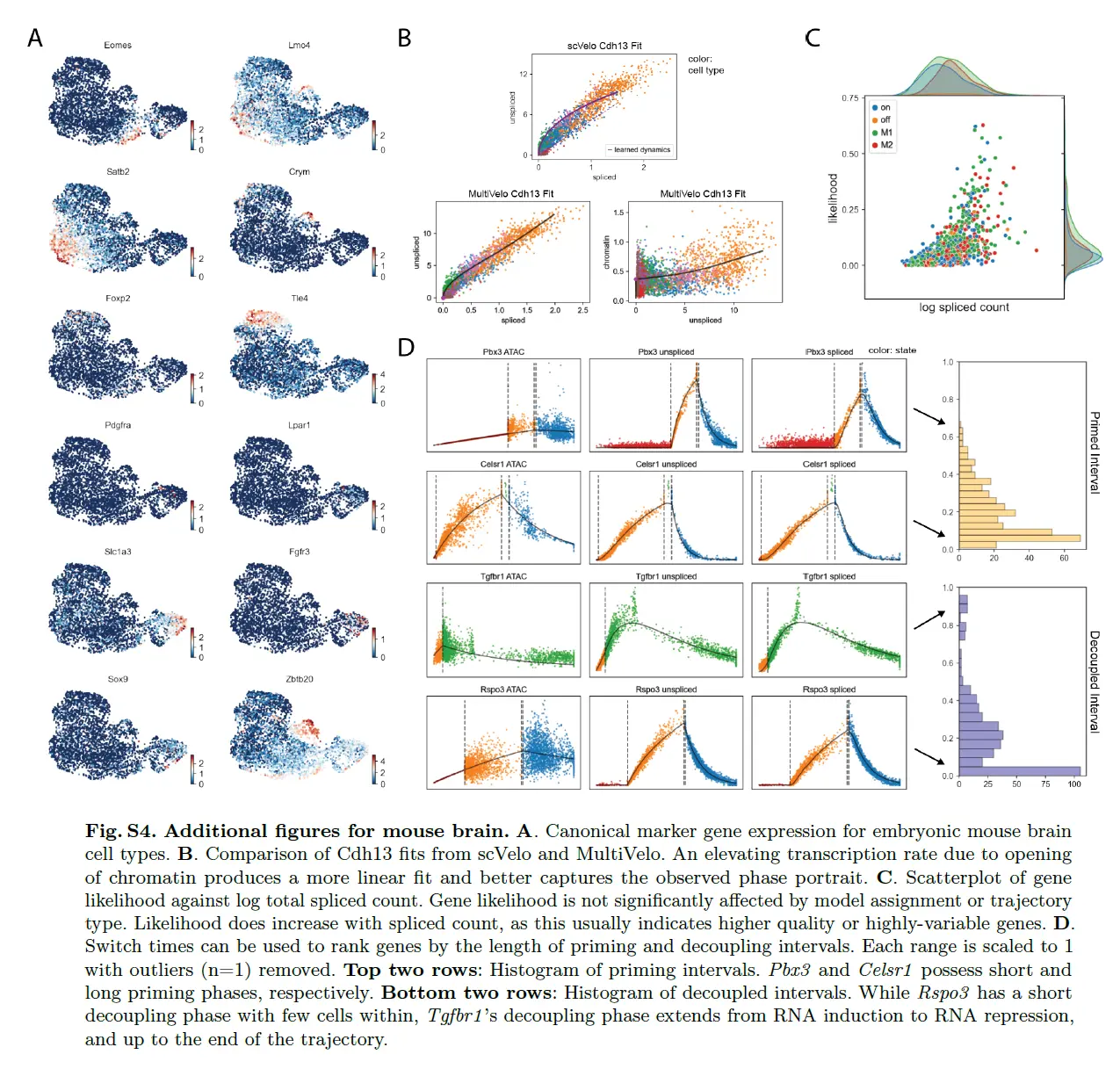
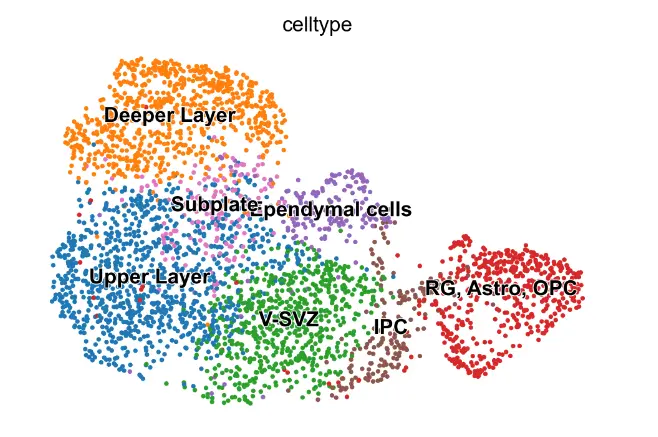
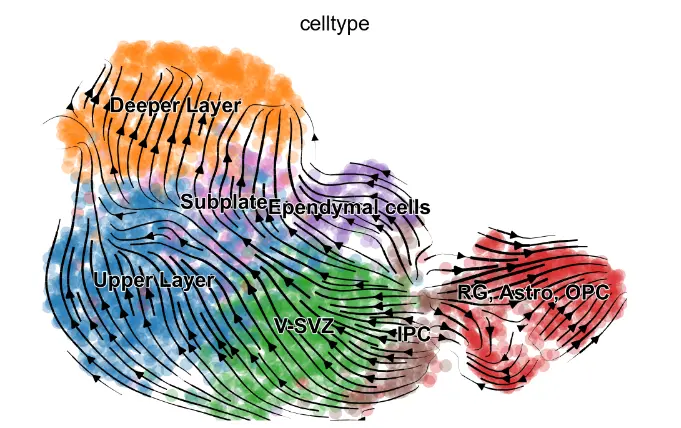
首先将 MultiVelo 应用于来自胚胎小鼠大脑 (E18) 的 10X Multiome 数据。 MultiVelo 准确拟合了观察到的染色质可及性、未剪接的前 mRNA 和剪接的 mRNA 计数在整个脑细胞群中,识别出 426 个基因,其模式极有可能符合模型。 MultiVelo 推断的所得速度向量和潜在时间值准确地恢复了哺乳动物皮层发育的已知轨迹。 具体来说,外脑室下区 (OSVZ) 中的径向神经胶质 (RG) 细胞会产生神经元、星形胶质细胞和少突胶质细胞。 皮质层在神经元迁移过程中以由内而外的方式形成,新生细胞移动到上层,老细胞留在更深的层。 RG细胞可以分裂成作为神经干细胞的中间祖细胞(IPC),并在不同层进一步产生各种成熟的兴奋性神经元。
与 scVelo 等仅使用 RNA 的模型相比,结合染色质可及性和基因表达提高了velocyto估计的准确性。 特别是,仅 RNA 模型预测了上层神经元内在生物学上不可信的回流。 细胞周期评分表明发育过程始于 RG 附近的循环 population,证实了 MultiVelo 推断的延迟时间。 MultiVelo 和 scVelo 使用类似的参数设置和估计算法,表明表观基因组数据提供了有关细胞过去和未来状态的重要附加信息,而不仅仅是转录组数据提供的信息。
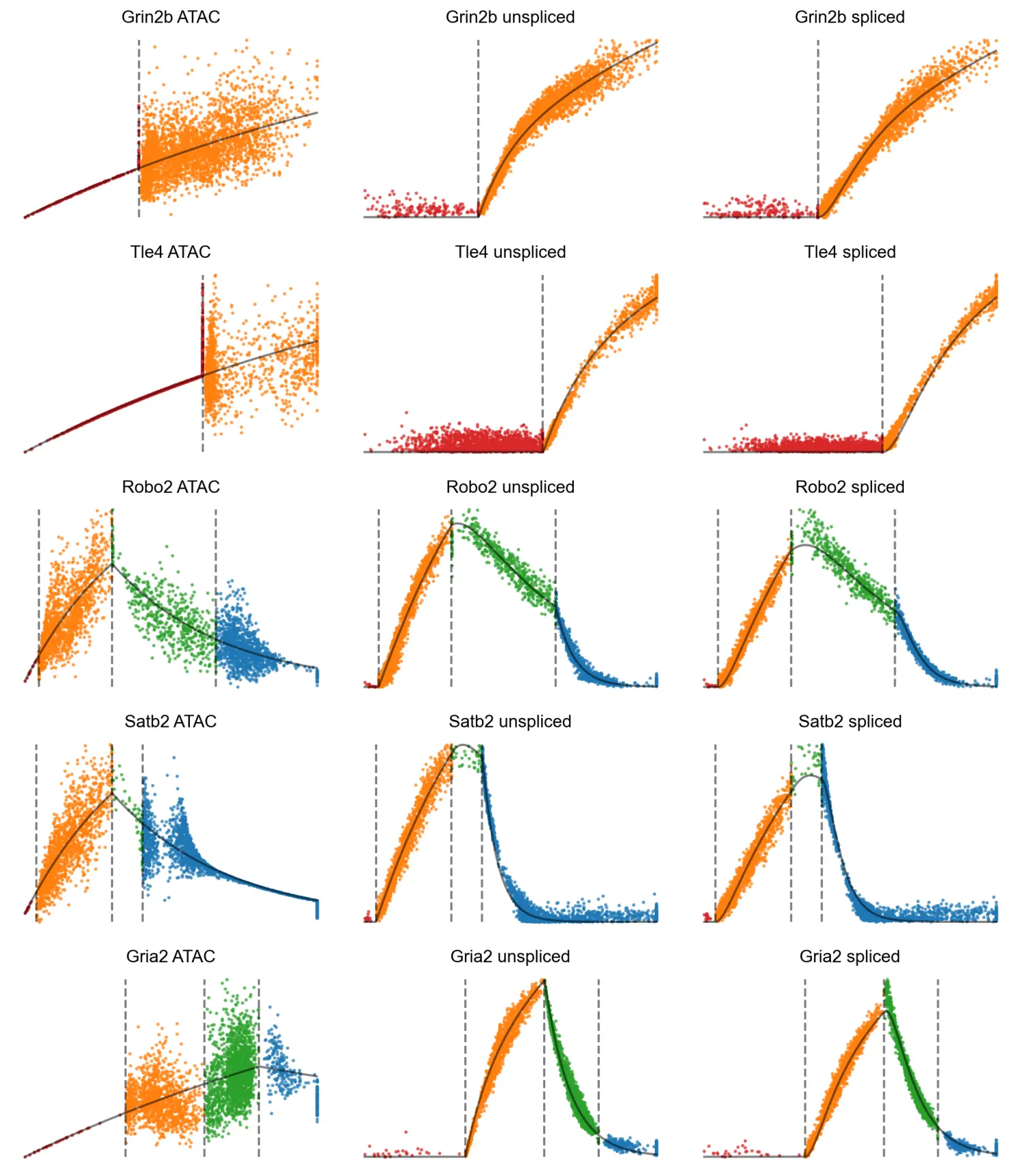
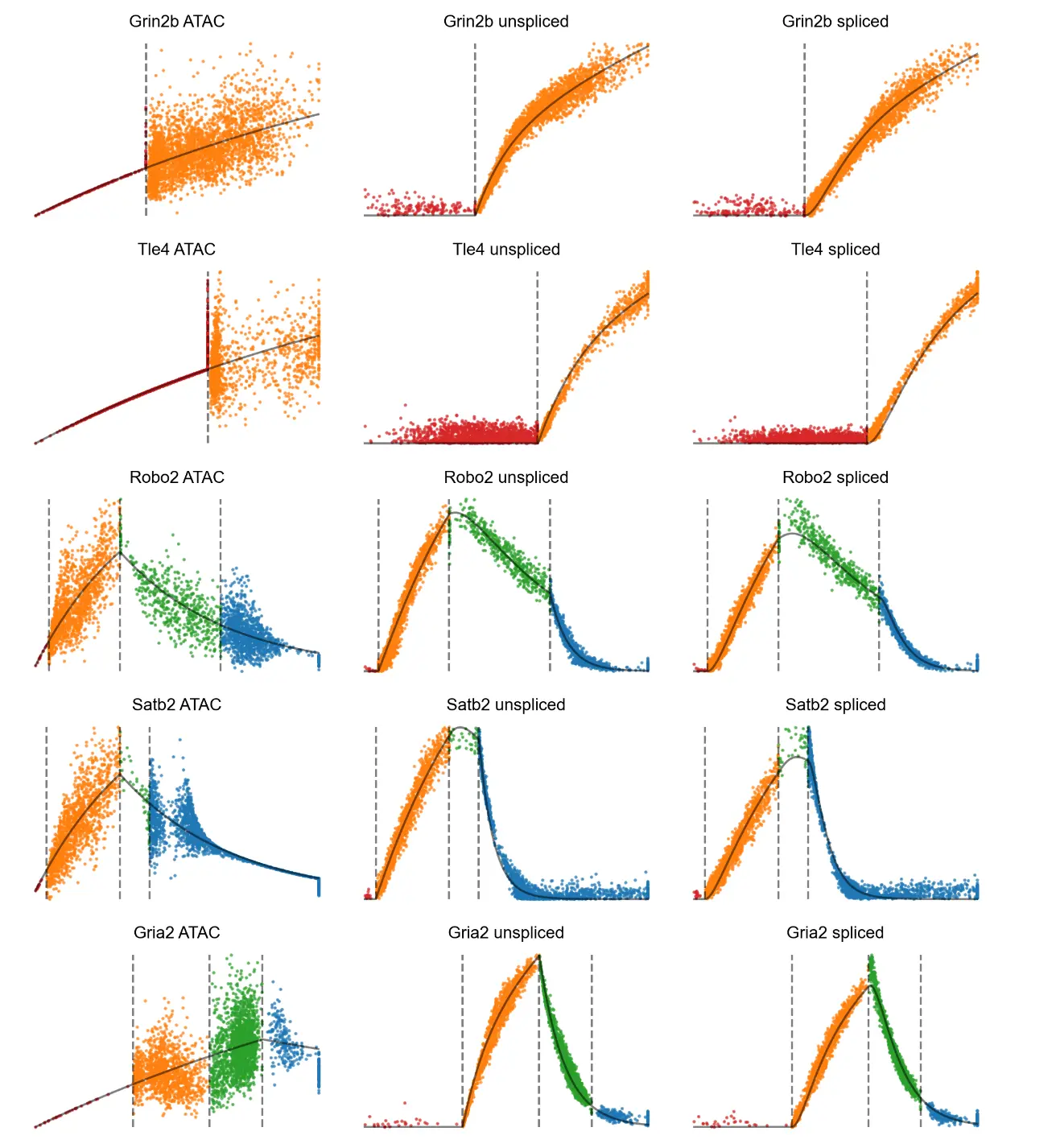
分析预计染色质可及性的添加最有助于区分染色质重塑和基因表达不同步的细胞状态,例如当基因的启动子和增强子开始打开但几乎没有发生转录时。两个明显的例子是 Eomes 和 Tle4,它们是 IPC 和深层神经元的典型标记。这些基因的 RNA 转录物仅在一种或两种特定细胞类型中高度表达。剩余的细胞密集地聚集在 (u, s) 相图的原点附近,使得 RNA 速度方法难以区分它们的相对顺序。然而,这些基因的染色质可及性在基因表达之前开始上升,揭示了仅从基因表达中看不到的逐渐变化。换句话说,结合染色质使我们能够推断出 3D 速度向量,指示每个细胞对每个基因的预测分化,比单独来自 RNA 的 2D 相图更好地解决细胞差异。
MultiVelo 确定了在此数据集中由模型 1 和模型 2 最好地描述的基因的清晰示例。 比较分配给模型 1 和模型 2 的基因的相图显示最大染色质可及性的时间存在明显差异,与模型预测一致。 模型 1 基因(如 Satb2)在转录诱导阶段(相图上的对角稳态线以上)达到最大染色质可及性,而模型 2 基因(如 Gria2)在转录抑制阶段(低于对角稳态线)的可及性最高 -状态线)。 在检查 c, u 和 c, s 的成对相图时,模型 1 和模型 2 之间的区别也很明显。 但是,不能通过单独检查 u, s 的相图中的 RNA 信息来区分模型; 区分需要来自染色质的额外信息。
进一步研究了模型 1 和模型 2 基因,看看它们是否具有任何特征。 Gene ontology(GO)分析表明,M2基因显着富集了与细胞周期相关的术语,如“细胞周期的正调控”、“有丝分裂细胞周期”和“细胞周期相变的调控”。 此外,模型 2 基因往往比模型 1 基因更早地在潜伏时间内达到最高剪接表达(p = 9 × 10−7,Wilcoxon 秩和单边检验)。 假设细胞可以使用模型 2 来快速、瞬时激活不需要维持表达的基因,而模型 1 可能对需要稳定表达的基因有用。
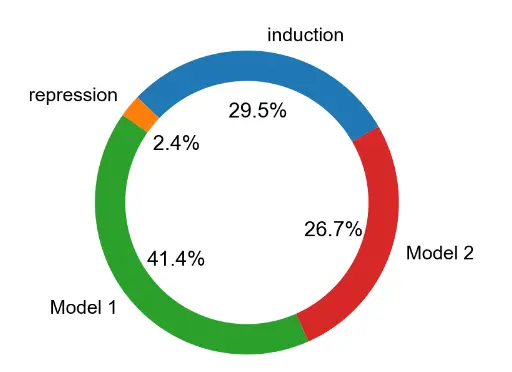
接下来研究了每种类型的基因表达动力学(仅诱导、仅抑制、模型 1 或模型 2)发生的频率。 大多数高度可变的基因同时显示诱导和抑制阶段(完整轨迹),对于只有部分轨迹的基因,仅诱导相图出现的频率高于仅抑制相图(29.5% 对 2.4% 的可变基因。Note,因为模型 1 和模型 2 在诱导阶段做出了相同的预测,无法区分模型 1 和模型 2 对于仅诱导基因。 1(41.4%的可变基因),而其余的最适合模型2(26.7%的可变基因)模型1更常见的事实与染色质状态变化通常先于mRNA表达变化的预期一致。
无论基因是否具有完整或部分动力学,MultiVelo 都适合描述其染色质可及性和基因表达动力学的三维轨迹的 ODE 参数。 通过对随时间变化的转录率进行建模,MultiVelo 能够更好地捕捉 RNA 相图中不同类型的曲率,而纯 RNA 模型无法捕捉到这种曲率差异。 具有不同模型分配和动力学的基因在可能性或总计数方面没有表现出显着差异,表明technical artifacts无法解释这些现象。
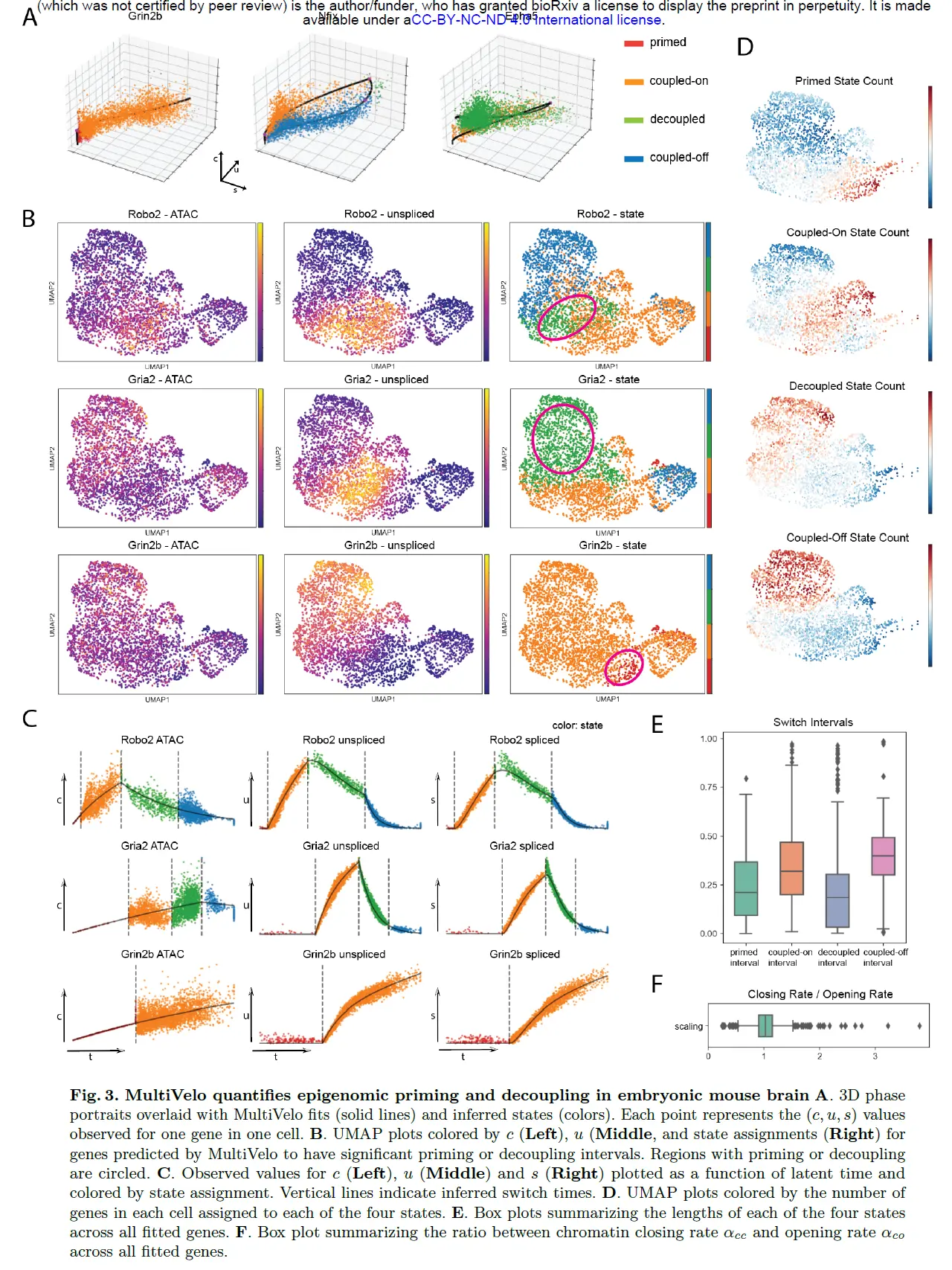
MultiVelo Identifies Epigenomic Priming and Decoupling in Embryonic Mouse Brain
MultiVelo 的一个重要的特性是它能够量化染色质可及性和分化细胞内基因表达之间的不一致和一致。 具体来说,MultiVelo 推断开关时间参数,这些参数确定每个基因处于四种可能状态(启动、耦合开启、解耦和耦合关闭)之一的间隔。 接下来研究了这些推断的状态和时间间隔是否可以准确地捕捉胚胎小鼠脑细胞中表观基因组和转录组变化之间的相互作用。
MultiVelo 识别了 10X Multiome 数据中四种状态中每一种的清晰示例。 例如,Grin2b 是一种仅诱导基因,其表达朝着神经元命运增加,因此仅预测了该基因的诱导状态——启动和耦合。 模型 1 基因 Nfix 的相图具有完整的轨迹形状,并标有所有四种状态。 相反,Epha5 是 Model 2 基因,它的可及性在整个时间范围内持续上升,没有观察到关闭阶段,所以它只占据耦合开启和解耦状态 。
通过在 UMAP 坐标上绘制可访问性 (c) 和表达式(u 和 s)并并排检查它们,可以定性地确认状态分配。在视觉上,观察到 c 和 u UMAP 图的颜色在状态分配耦合打开或耦合关闭时匹配,并且在分配状态启动或解耦时出现颜色差异。例如,Robo2 RNA 表达和染色质可及性之间的最大差异发生在圆圈区域,该区域预计处于解耦状态。 Robo2 是一个 Model 1 基因;染色质闭合开始后,表达保持在相对较高的水平,即使它的可及性已经经历了成熟神经元的下降。同样,Gria2 的可访问性与解耦状态的 RNA 不同。模型 2 基因 Gria2 的染色质可及性在转录诱导期之后继续增加。此外,基因 Grin2b 显示了染色质引发阶段的一个明显例子,在此期间染色质在 RNA 产生之前打开。
沿着每个基因的推断时间 t 绘制 c、u 和 s 能够详细检查状态转换。首先,Robo2 的 u(t) 和 s(t) 值在转录抑制阶段显示出两个拐点,对应于从耦合到解耦状态和从解耦到耦合关闭状态的转变。这种模式表明染色质关闭和转录抑制的不同影响在 u(t) 和 s(t) 中可见。换句话说,MultiVelo 预测对于 Robo2,染色质关闭会降低整体转录率,因为 RNA 水平在染色质转换后立即开始下降。随后转录率从正值到零的转换导致第二个拐点,导致 RNA 表达的更快速下调。 Gria2 的 c(t)、u(t) 和 s(t) 的图显示出相反的趋势:即使在切换到转录抑制后,c 仍继续上升,导致 c 和 u 在解耦状态期间向相反的方向移动.在 Grin2b 的长启动阶段,c(t) 开始上升,而 u(t) 和 s(t) 保持为零 。
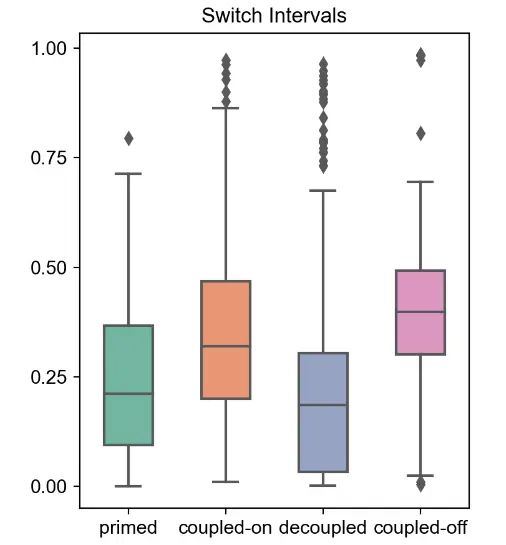
由于 MultiVelo 适合每个基因的速率和转换时间参数,分析提供了观察基因调控总体趋势的机会。首先,为了确定不同基因的状态是否在时间上是协调的,计算了每个细胞每个状态中高似然基因的数量。确实存在通过神经元cluster的级联状态转换;每个细胞的多个基因通常同时处于启动或去耦状态。其次,寻找转换时间和速率参数的趋势。将每个基因的诱导/抑制周期置于 0 到 1 之间的时间尺度上,发现耦合开启和耦合关闭状态比启动和解耦状态占基因表达过程的更大比例。这是有道理的,因为即使基因在两种模式之间经历某种程度的解耦和时间滞后,染色质可及性和基因表达仍应普遍相关。中位数启动间隔长度为总时间的 21%,中位数解耦间隔长度为总时间的 19%。此外,可以通过启动和解耦间隔的时间对基因进行排序,以找到可访问性和表达之间不一致的例子。此外,发现染色质通常以相似的速度打开和关闭:推断的染色质关闭率 (αcc) 和染色质打开率 (αco) 之间的中值比率几乎正好是 1。
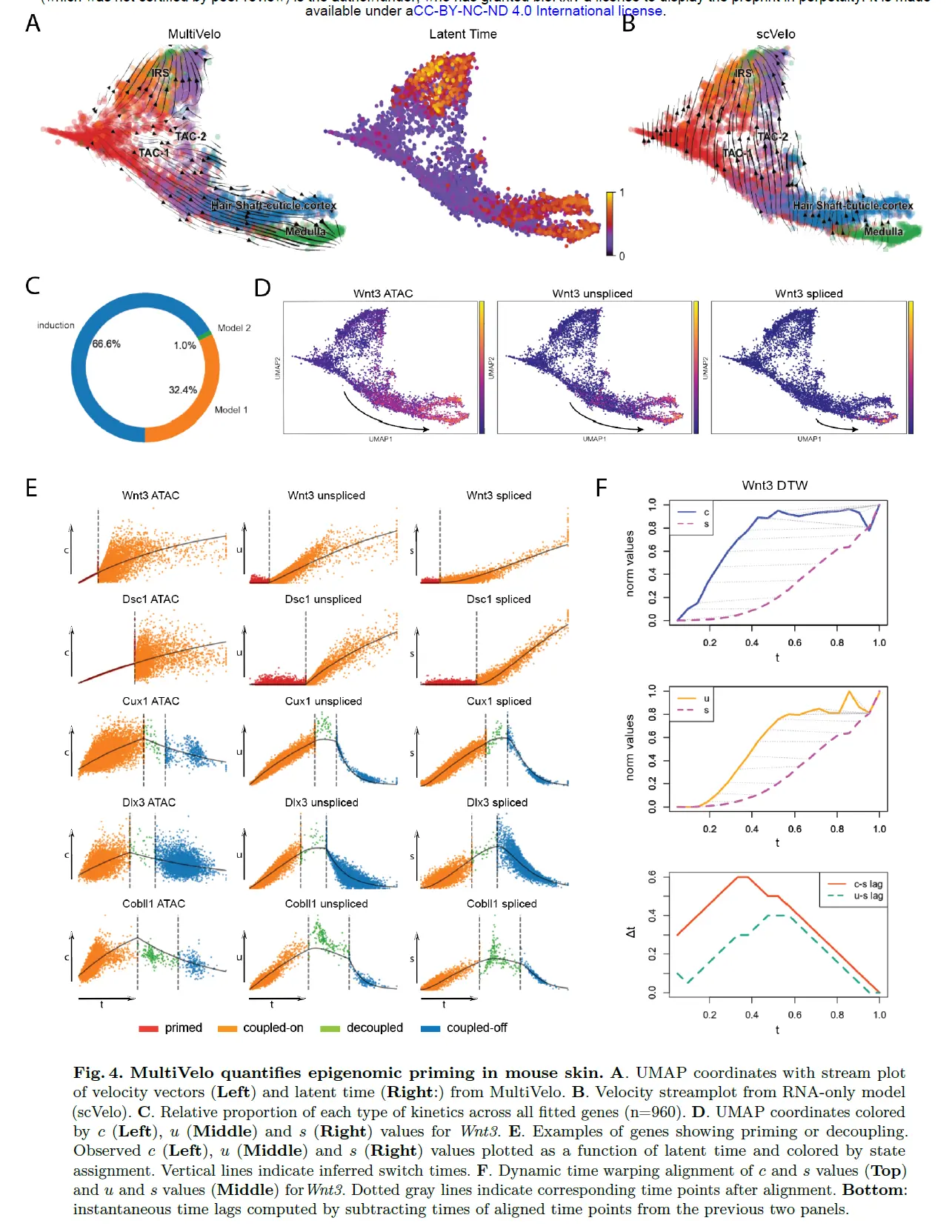
MultiVelo Quantifies Epigenomic Priming in SHARE-seq Data from Mouse Hair Follicle
最近的一项研究使用 SHARE-seq 来研究毛囊组织中转运放大细胞 (TAC) 的快速增殖,这些细胞会产生几种成熟的效应细胞,包括内根鞘 (IRS) 和毛干层:角质层、皮质 层和髓质。 当应用于该数据集时,MultiVelo 正确识别了从 TAC 到 IRS 和毛干细胞的分化方向,与最初论文中报告的扩散图分析一致。 潜伏时间预测 TAC 是根细胞——与生物学预期一致——而单独使用 RNA 的速度分析未能捕捉到毛干分化方向。 与 mouse brai相比,在该数据集中观察到明显更多的仅诱导基因和更少的模型 2 基因。
最初的 SHARE-seq 论文的主要结果之一是鉴定了启动子和增强子染色质可及性预示基因表达的基因,作者将这种现象称为“染色质潜力”。这种现象最明显的例子是 Wnt3,它编码旁分泌信号分子,在控制头发生长方面很重要。事实上,UMAP 图按可及性着色,未剪接和剪接的 mRNA 表达显示出明显的时间延迟。接下来检查了 SHARE-seq 论文中确定的其他基因。拟合模型显示 MultiVelo 忠实地捕捉了每个基因的动态,并清楚地说明了启动和去耦区域。例如,Wnt3 和 Dsc1 显示仅诱导模式和开始时的启动状态,而 Cux1、Dlx3 和 Cobll1 具有诱导和抑制状态 中间有一个短的解耦期。
为了进一步量化可及性、未拼接表达和拼接表达之间的时间关系,使用动态时间扭曲 (DTW) 来对齐每个分子层的时间序列值。 DTW 非线性扭曲两个时间序列以最大化它们的相似性并识别可能的滞后相关性。 Wnt3 上的 DTW 结果表明,最佳扭曲函数在时间上向前映射 c 时间序列上的每个点,与基因表达之前的染色质可及性一致。未拼接和拼接的表达显示出相似的模式,但延迟时间较短。由于 DTW 将前面曲线上的每个时间点映射到后面曲线上的时间点,因此可以通过减去匹配点的时间来计算每个时间点的时滞。该分析表明,c 和 s 之间的延迟以及 u 和 s 之间的延迟在整个观察时间内都保持为正值。此外,在整个观测范围内,c 和 s 之间的延迟比 u 和 s 之间的延迟长,最大 c 和 s 延迟达到 0.6(在总时间范围 1 之外)。
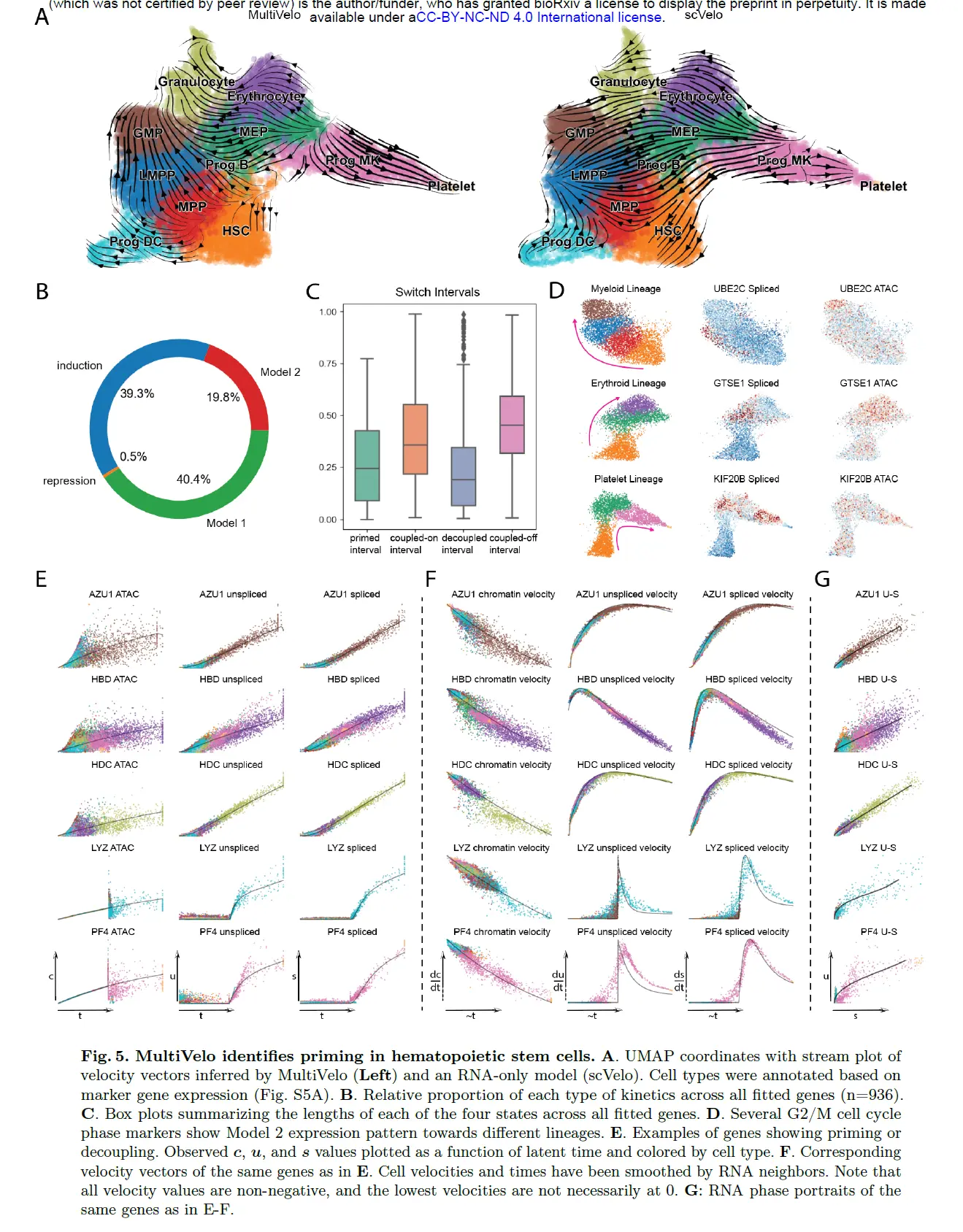
MultiVelo Reveals Early Epigenomic and Transcriptomic Changes in Human Hematopoietic Stem and Progenitor Cells
造血祖细胞由干细胞样细胞群组成,这些细胞群在进入更多谱系受限状态时,可快速且持续地分化为各种中等和成熟血细胞类型,自我更新能力逐渐降低
将纯化的人类 CD34+ 细胞培养了 7 天,然后使用 10X Multiome 平台对其进行测序。 使用单核 RNA-seq 和 ATAC-seq 数据过滤后获得了 11,605 个高质量细胞。 使用先前描述的标记基因,确定了类似于许多早期血液发育群体的cluster,包括 HSC、多能祖细胞 (MPP)、淋巴引发的多能祖细胞 (LMPP)、粒细胞-巨噬细胞祖细胞 (GMP) 和巨核细胞- 红细胞祖细胞 (MEP)。 还确定了类似于早期粒细胞、红细胞、树突细胞 (DC) 和血小板的cluster。
血细胞分化是用 RNA 速度建模的一个具有挑战性的系统,但发现结合染色质信息显着提高了预测细胞方向的局部一致性和生物学准确性。 相比之下,仅从 RNA 推断的速度向量并不能准确反映 HSPC 的已知分化层次。 与小鼠大脑一样,MultiVelo 预测模型 1 在该数据集中比模型 2 更常见; 仅归纳是第三个最常见的基因类。 观察到的启动和解耦区间的中值长度比耦合阶段的短。 这些模式与我们在小鼠大脑数据集中观察到的一致,表明可能存在共同的潜在生物学机制 。
与小鼠大脑数据集一样,HSPC 数据集中的模型 2 基因显着丰富了与细胞周期相关的 GO terms。 terms“有丝分裂细胞周期的调节”、“有丝分裂中期/后期转变的调节”和“有丝分裂姐妹染色单体分离的调节”都富集在模型2基因中,FDR < 0.002。 如果我们检查髓系、红细胞系和血小板谱系的不同轨迹,许多 G2/M 期标记基因 18 显示清晰的模型 2 模式,在表达开始下降后染色质可及性最高 。
进一步研究了模型 1 和模型 2 基因的组蛋白修饰谱是否不同。 由于可以使用 FACS 对经典定义的 HSPC 亚群进行分类,因此我们分析中的某些细胞亚群可以获得大量 ChIP-seq 数据。 使用这些大量数据集,我们比较了 FACS 纯化的 HSC 中 H3K4me3、H3K4me1 和 H3K27ac 的水平,染色质可及性峰值与模型 1 和模型 2 基因相关。 我们发现模型 2 基因显示出显着更高的 H3K4me3(p = 0.016,单侧 Wilcoxon 秩和检验),这是活跃启动子的标志。 相比之下,模型 1 基因显示出稍高的 H3K4me1 (p = 0.097),这是一种启动的增强子标记。 两种模型在 HSC 中都显示出相似的 H3K27ac(一种活性增强子标记物)(p = 0.48)。
MultiVelo 拟合的基因模型揭示了许多启动的例子。 几种终末细胞类型特异性标记物显示出仅诱导动力学,染色质可及性增加,随后基因表达增加(GMP 中的 AZU1、红细胞中的 HBD、粒细胞中的 HDC、DC 祖细胞中的 LYZ 和巨核细胞 (MK) 祖细胞中的 PF4) 方向)。 在 HSPC 中,我们再次看到一些明显的长启动期示例,例如 LYZ 和 PF4
绘制Velocyto使我们能够更详细地检查局部染色质和 RNA 趋势。 虽然染色质在这些基因开始时显示出最大的潜力(最高速度),但对于 RNA,干细胞群(如 HSC、MPP、MEP 和 GMP)在向一个谱系的分化过程中显示出增加的潜力。 更多分化的细胞类型失去了保持这种潜力的能力,并逐渐接近平衡(零速度),即使表达仍在有所增加。 请注意,即使整体表达式升高且速度保持为正,局部加速度仍可以切换符号。 由于染色质和 mRNA 的联合数学建模,MultiVelo 能够捕获关于分化方向和速率的如此丰富的信息。 添加染色质显着丰富了从 RNA 中可用的信息,这可以通过检查仅 RNA 相图看出。
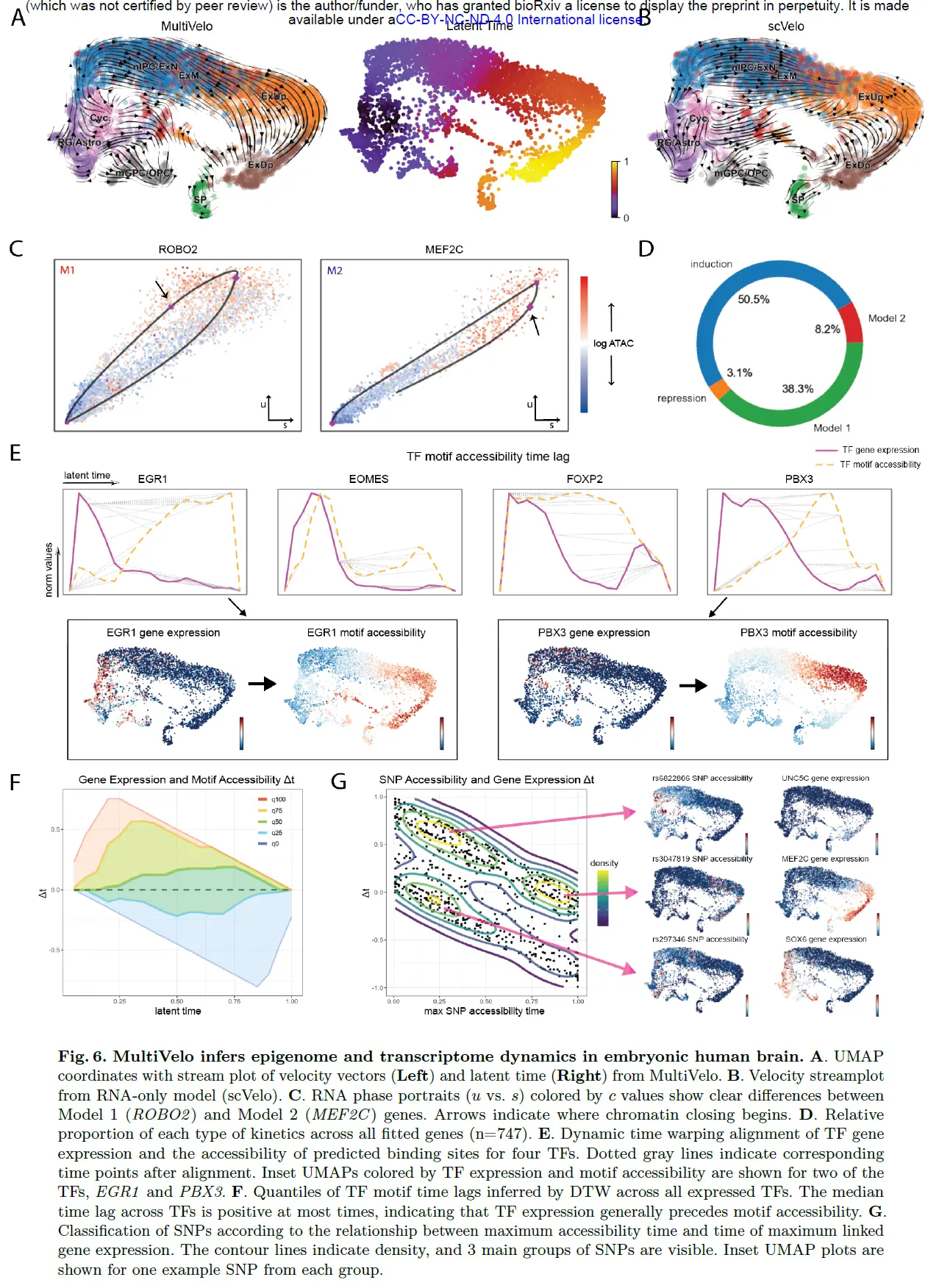
MultiVelo Relates Transcription Factors, Polymorphic Sites, and Gene Expression in Developing Human Brain
接下来将 MultiVelo 应用到最近发布的来自开发人类皮层的 10X Multiome 数据集。 与胚胎小鼠大脑数据集一样,MultiVelo 推断出的速度向量与已知的脑细胞发育模式一致。 MultiVelo 正确地推断出放射状胶质细胞附近的循环细胞群是潜伏时间最早的细胞类型。 相比之下,在没有染色质信息的情况下推断的速度向量预测了中间祖细胞和上层兴奋性神经元的不协调回流。
与小鼠大脑数据集一样,确定了模型 1 和模型 2 基因的清晰示例,尽管在人类数据集中预测遵循模型 2 的基因较少。 有趣的是,模型 2 基因 MEF2C 被仅 RNA 模型预测为具有主要的抑制相,可能是因为 u - s 相图的“宽度”很窄。然而,染色质信息的添加允许正确的预测该基因同时具有诱导期和抑制期。
MultiVelo 的一个关键优势是它能够将细胞置于从染色质和表达数据推断出的潜伏时间尺度上。我们推断,潜伏时间可以识别基因座的表达和可及性之间的时间滞后,而不仅仅是那些紧邻基因的基因座。例如,潜伏时间可用于计算转录因子 (TF) 的表达与其结合位点的可及性之间的时间长度。为此,我们使用 chromVar40 计算每个细胞的峰与每个 TF 的结合位点的总可及性,子集仅针对数据集中可变表达的 TF。然后我们使用动态时间扭曲 (DTW) 将每个 TF 的时间序列表达与其绑定站点的可访问性对齐。这揭示了一个一致的模式,其中转录因子最高 RNA 表达的时间先于相应的下游靶标的高可及性时间。由 TF 表达和结合位点可访问性着色的 UMAP 图在视觉上证实了这种模式。所有表达的 TF 的中位时间滞后为正,表明在大多数情况下 TF 表达先于结合位点可访问性。如果没有额外的数据,我们无法最终确定这些时间滞后背后的机制。然而,转录后和翻译后调节、影响染色质重塑复合物活性的因素以及细胞间信号传导都可能导致这种现象。
MultiVelo 推断的潜伏时间也可用于将疾病相关变异基因座的染色质可及性与附近基因的表达联系起来。我们收集了 6968 个单核苷酸多态性 (SNP) 及其关联基因的列表,这些基因与精神疾病(包括双相情感障碍和精神分裂症)的全基因组关联研究有关。我们进一步将这些 SNP 子集到与我们模型拟合的基因相关的重叠染色质可及性峰,总共 757 个 SNP。许多这些变异发生在神经元转录因子和其他发育重要基因附近。然后我们计算了 400 b.p. 每个细胞的染色质可及性。窗口以每个 SNP 为中心。使用 MultiVelo 的潜伏时间,我们确定了每个 SNP 的最大可达性时间以及 SNP 可达性与其连锁基因的最大表达之间的时间滞后。该分析揭示了 3 个主要的 SNP 组,根据它们的最大可接近性是在潜伏时间的早期还是晚期以及相关基因表达之前或之后进行区分。 SNP 可及性和相关基因表达的 UMAP 图证实这些 SNP 组具有不同的性质。这些分组对于理解 SNP 的功能很重要;例如,与完全分化的细胞相比,仅在潜伏期早期才可接近的 SNP 在发育细胞中可能发挥更大的作用。同样,可接近性先于基因表达的 SNP 比可接近性落后的 SNP 更有可能参与调节其表达。
Discussion
总之,MultiVelo 准确地恢复了细胞谱系,并量化了染色质可及性和基因表达暂时不同步的引发和解偶联间隔的长度。 我们的模型准确地拟合了来自胚胎小鼠大脑、小鼠背部皮肤、胚胎人类大脑和人类造血干细胞的单细胞多组学数据集。 此外,我们的模型确定了两类基因,它们在染色质关闭和转录抑制的相对顺序上有所不同,我们在我们研究的所有组织中找到了这两种机制的清晰例子。 我们预计 MultiVelo 将深入了解一系列生物环境中基因表达的表观基因组调控,包括正常细胞分化、重编程和疾病。
Method(又到了数学的时间)
Previous Approaches: RNA velocity

Differential Equation Model of Gene Expression Incorporating Chromatin Accessibility


Model Likelihood

Parameter Estimation and Latent Time Inference by Expectation Maximization
Both the cell times t and the ODE parameters are unknown, so we perform expectation-maximization to simultaneously infer them. The E-step involves determining the expected value of latent time for each cell given the current best estimate of the ODE parameters. Because inverting the three-dimensional ODEs analytically is not straightforward, we perform this time estimation by finding the time whose ODE prediction is nearest each data point, selecting the time from a vector of uniformly spaced time points (see Implementation Detail section). In the M-step, we find the ODE parameters that maximize the data likelihood (equivalent to minimizing MSE) given the current time estimates for each cell. We use the Nelder-Mead simplex algorithm to minimize MSE.
Model Pre-Determination and Distinguishing Genes with Partial and Complete Dynamics

Parameter Initialization(最难的部分)


示例代码
加载
import os
import scipy
import numpy as np
import pandas as pd
import scanpy as sc
import scvelo as scv
import multivelo as mv
import matplotlib.pyplot as plt
from IPython.core.display import display, HTML
display(HTML("<style>div.output_scroll { height: 50em; }</style>"))
display(HTML("<style>.container { width:80% !important; }</style>"))
scv.settings.verbosity = 3
scv.settings.presenter_view = True
scv.set_figure_params('scvelo')
pd.set_option('display.max_columns', 100)
pd.set_option('display.max_rows', 200)
np.set_printoptions(suppress=True)
Reading in unspliced and spliced counts
adata_rna = scv.read("velocyto/10X_multiome_mouse_brain.loom", cache=True)
adata_rna.obs_names = [x.split(':')[1][:-1] + '-1' for x in adata_rna.obs_names]
adata_rna.var_names_make_unique()
sc.pp.filter_cells(adata_rna, min_counts=1000)
sc.pp.filter_cells(adata_rna, max_counts=20000)
# Top 1000 variable genes are used for downstream analyses.
scv.pp.filter_genes(adata_rna, min_shared_counts=10)
scv.pp.normalize_per_cell(adata_rna)
scv.pp.filter_genes_dispersion(adata_rna, n_top_genes=1000)
# Load cell annotations
cell_annot = pd.read_csv('cell_annotations.tsv', sep='\t', index_col=0)
adata_rna = adata_rna[cell_annot.index,:]
adata_rna.obs['celltype'] = cell_annot['celltype']
# We subset for lineages towards neurons and astrocytes.
adata_rna = adata_rna[adata_rna.obs['celltype'].isin(['RG, Astro, OPC',
'IPC',
'V-SVZ',
'Upper Layer',
'Deeper Layer',
'Ependymal cells',
'Subplate'])]
adata_rna
View of AnnData object with n_obs × n_vars = 3653 × 1000
obs: 'n_counts', 'initial_size_spliced', 'initial_size_unspliced', 'initial_size', 'celltype'
var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'gene_count_corr', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
layers: 'ambiguous', 'matrix', 'spliced', 'unspliced'
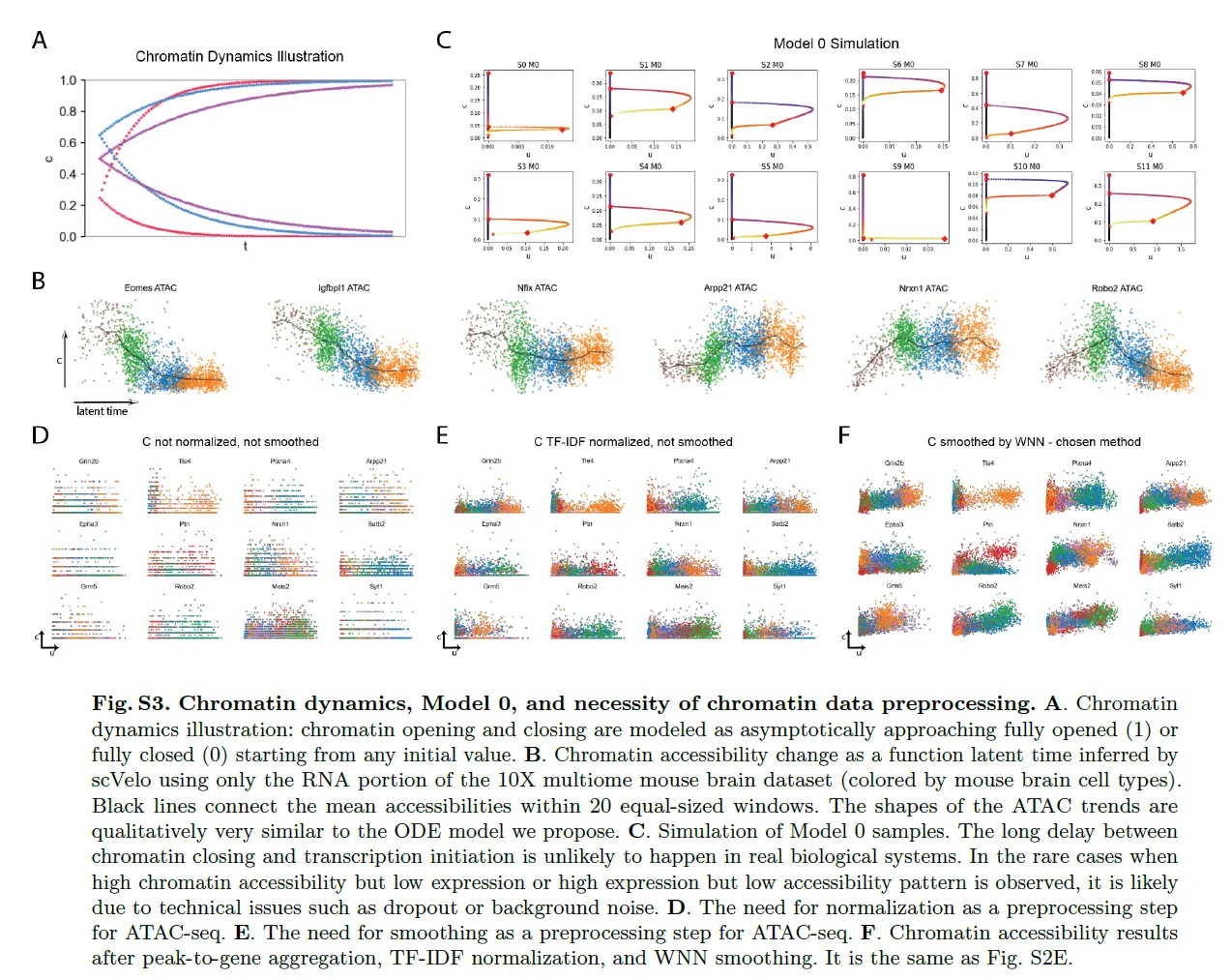
Preprocessing the ATAC counts
adata_atac = sc.read_10x_mtx('outs/filtered_feature_bc_matrix/', var_names='gene_symbols', cache=True, gex_only=False)
adata_atac = adata_atac[:,adata_atac.var['feature_types'] == "Peaks"]
# We aggregate peaks around each gene as well as those that have high correlations with promoter peak or gene expression.
# Peak annotation contains the metadata for all peaks.
# Feature linkage contains pairs of correlated genomic features.
adata_atac = mv.aggregate_peaks_10x(adata_atac,
'outs/peak_annotation.tsv',
'outs/analysis/feature_linkage/feature_linkage.bedpe',
verbose=True)
# Let's examine the total count distribution and remove outliers.
plt.hist(adata_atac.X.sum(1), bins=100, range=(0, 100000));
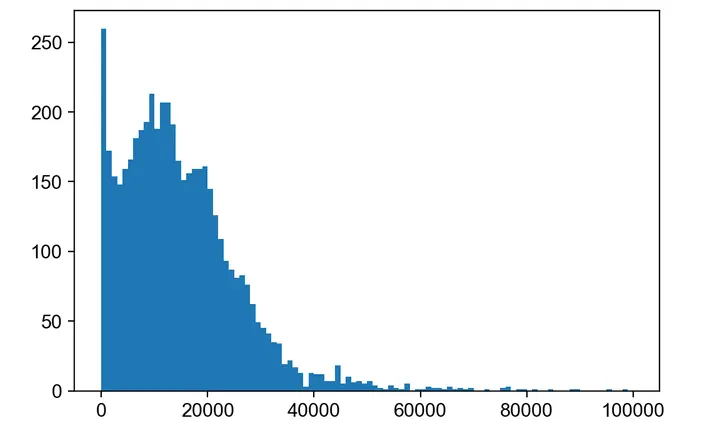
sc.pp.filter_cells(adata_atac, min_counts=2000)
sc.pp.filter_cells(adata_atac, max_counts=60000)
# We normalize aggregated peaks with TF-IDF.
mv.tfidf_norm(adata_atac)
Finding shared barcodes and features between RNA and ATAC
shared_cells = pd.Index(np.intersect1d(adata_rna.obs_names, adata_atac.obs_names))
shared_genes = pd.Index(np.intersect1d(adata_rna.var_names, adata_atac.var_names))
len(shared_cells), len(shared_genes)
###(3365, 936)
# We reload in the raw data and continue with a subset of cells.
adata_rna = scv.read("velocyto/10X_multiome_mouse_brain.loom", cache=True)
adata_rna.obs_names = [x.split(':')[1][:-1] + '-1' for x in adata_rna.obs_names]
adata_rna.var_names_make_unique()
adata_rna = adata_rna[shared_cells, shared_genes]
adata_atac = adata_atac[shared_cells, shared_genes]
scv.pp.normalize_per_cell(adata_rna)
scv.pp.log1p(adata_rna)
scv.pp.moments(adata_rna, n_pcs=30, n_neighbors=50)
adata_rna.obs['celltype'] = cell_annot.loc[adata_rna.obs_names, 'celltype']
adata_rna.obs['celltype'] = adata_rna.obs['celltype'].astype('category')
# Reorder the categories for color consistency with the manuscript.
all_clusters = ['Upper Layer',
'Deeper Layer',
'V-SVZ',
'RG, Astro, OPC',
'Ependymal cells',
'IPC',
'Subplate']
adata_rna.obs['celltype'] = adata_rna.obs['celltype'].cat.reorder_categories(all_clusters)
scv.tl.umap(adata_rna)
scv.pl.umap(adata_rna, color='celltype')
Smoothing gene aggregagted peaks by neighbors
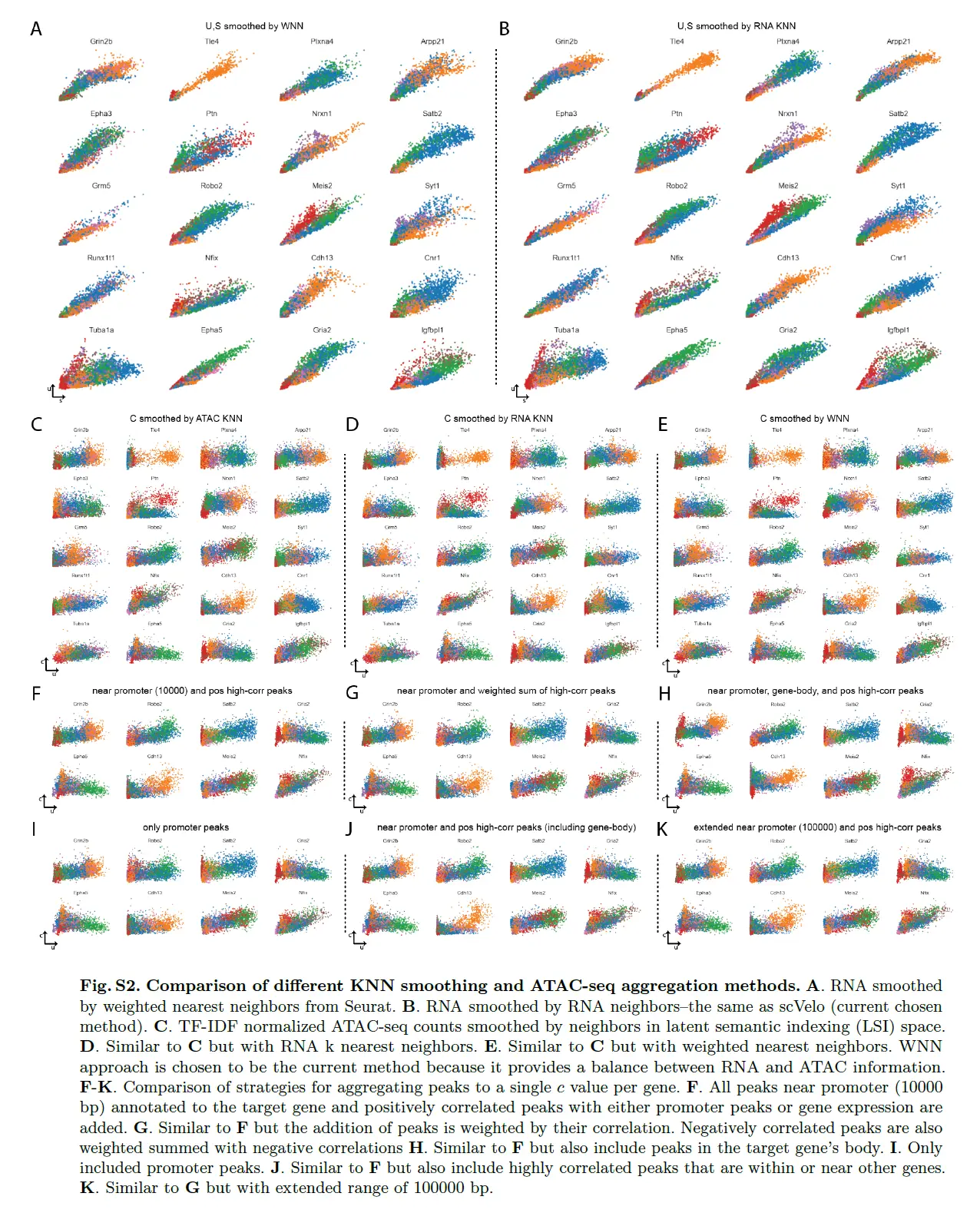
# Read in Seurat WNN neighbors.
nn_idx = np.loadtxt("seurat_wnn/nn_idx.txt", delimiter=',')
nn_dist = np.loadtxt("seurat_wnn/nn_dist.txt", delimiter=',')
nn_cells = pd.Index(pd.read_csv("seurat_wnn/nn_cells.txt", header=None)[0])
# Make sure cell names match.
np.all(nn_cells == adata_atac.obs_names)
mv.knn_smooth_chrom(adata_atac, nn_idx, nn_dist)
adata_atac
AnnData object with n_obs × n_vars = 3365 × 936
obs: 'n_counts'
layers: 'Mc'
obsp: 'connectivities'
Running multi-omic dynamical model
MultiVelo incorporates chromatin accessibility information into RNA velocity and achieves better lineage predictions.
The detailed argument list can be shown with "help(mv.recover_dynamics_chrom)".
# This will take a while.
adata_result = mv.recover_dynamics_chrom(adata_rna,
adata_atac,
max_iter=5,
init_mode="invert",
verbose=False,
plot=False,
parallel=True,
save_plot=False,
rna_only=False,
fit=True,
extra_color_key='celltype'
)
# Save the result for use later on
adata_result.write("multivelo_result.h5ad")
adata_result = sc.read_h5ad("multivelo_result.h5ad")
mv.pie_summary(adata_result)
Screenshot 2021-12-21 at 11-40-03 Notebook on nbviewer.png
mv.switch_time_summary(adata_result)
mv.likelihood_plot(adata_result)

By default, the velocity genes used for velocity graph is determined as those whose likelihoods are above 0.05. They can be reset with "mv.set_velocity_genes" function upon examining the distributions of variables above if needed.
Computing velocity stream and latent time
mv.velocity_graph(adata_result)
mv.latent_time(adata_result)
mv.velocity_embedding_stream(adata_result, basis='umap', color='celltype')
scv.pl.scatter(adata_result, color='latent_time', color_map='gnuplot', size=80)
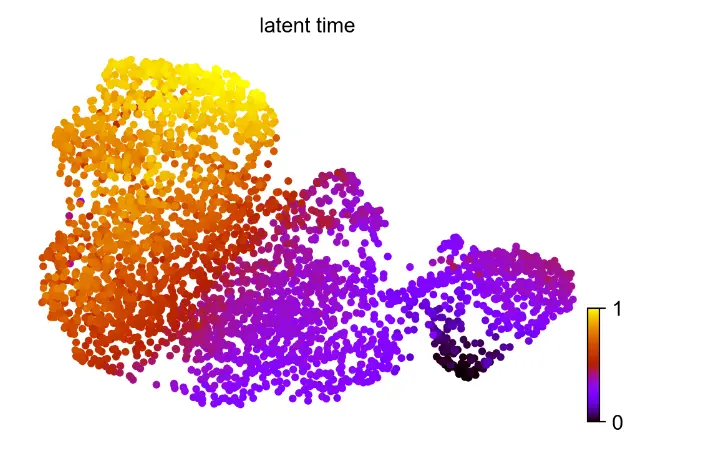
Let's examine some example genes
gene_list = ['Grin2b', 'Tle4', 'Robo2', 'Satb2', 'Gria2']
We can plot accessbility and expression against gene time
# Accessibility/expression by gene time, colored by the four potential states.
# The solid black curve indicates anchors.
mv.dynamic_plot(adata_result, gene_list, color_by='state', axis_on=False, frame_on=False)
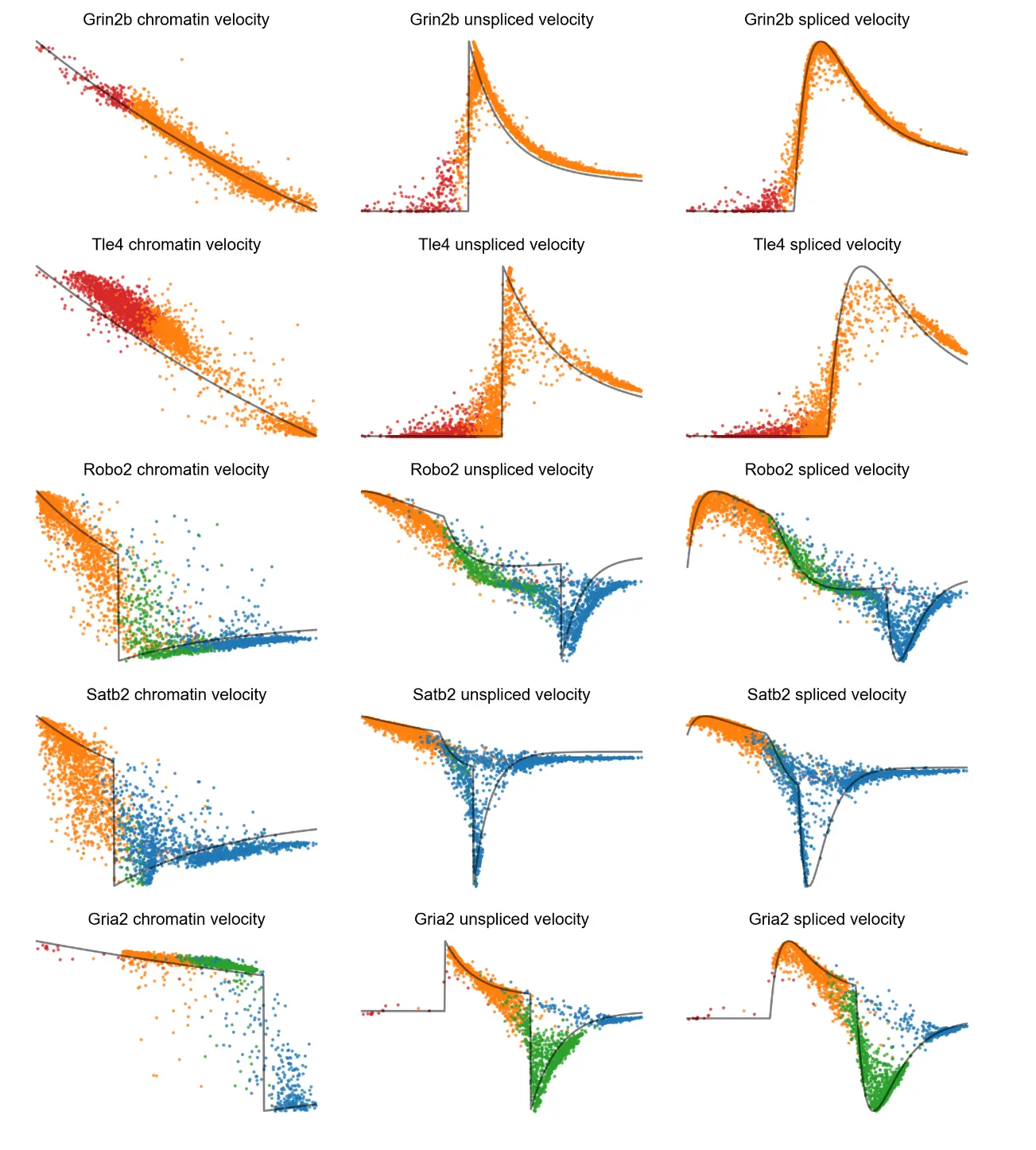
We can plot velocity against gene time
# Velocity by gene time, colored by the four potential states.
# The solid black curve indicates anchors.
mv.dynamic_plot(adata_result, gene_list, color_by='state', by='velocity', axis_on=False, frame_on=False)
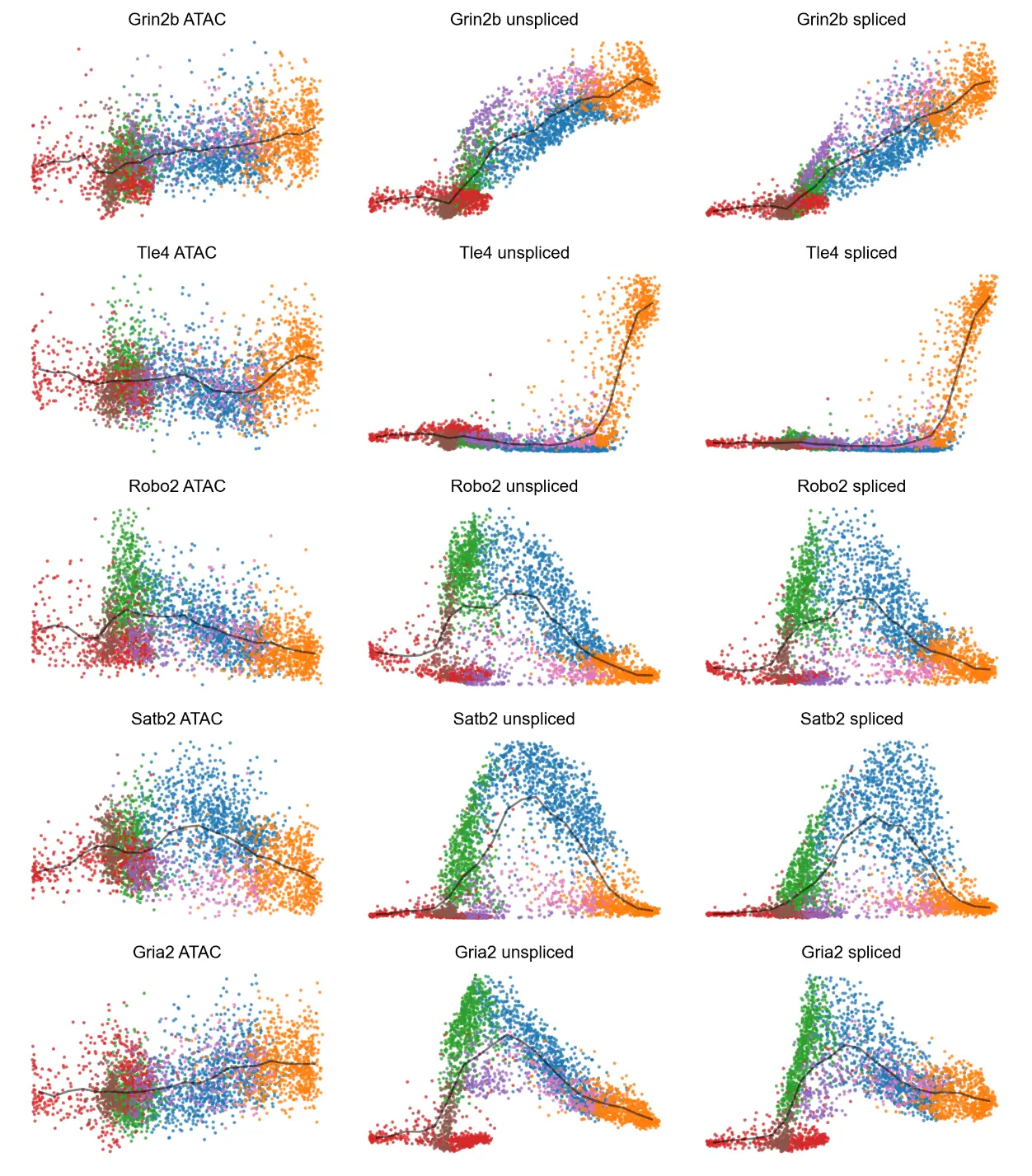
We can examine accessibility and expression against globally shared latent time
# Accessibility/expression by global latent time, colored by cell type assignments.
# The solid black curve indicates the mean.
mv.dynamic_plot(adata_result, gene_list, color_by='celltype', gene_time=False, axis_on=False, frame_on=False)
Phase portraits on the unspliced-spliced, chromatin-unspliced, or 3-dimensional planes can be plotted
# Unspliced-spliced phase portraits, colored by celltype.
mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='us', axis_on=False, frame_on=False)

# Unspliced-spliced phase portraits, colored by log chromatin accessibility.
# title_more_info shows more information in each subplot title: model, direction, and likelihood.
mv.scatter_plot(adata_result, gene_list, color_by='c', by='us', cmap='coolwarm', title_more_info=True, axis_on=False, frame_on=False)
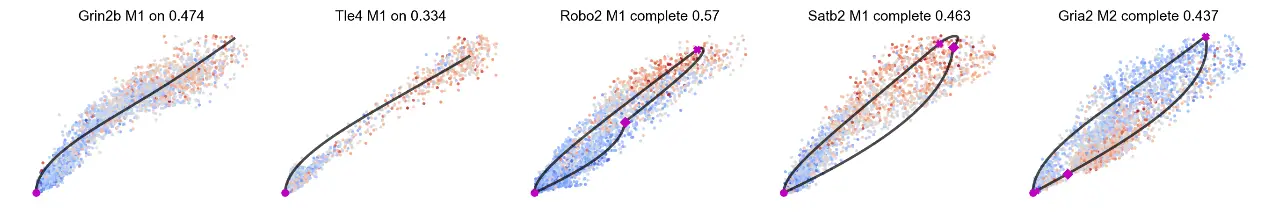
# Chromatin-unspliced phase portraits, colored by celltype.
mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='cu', axis_on=False, frame_on=False)
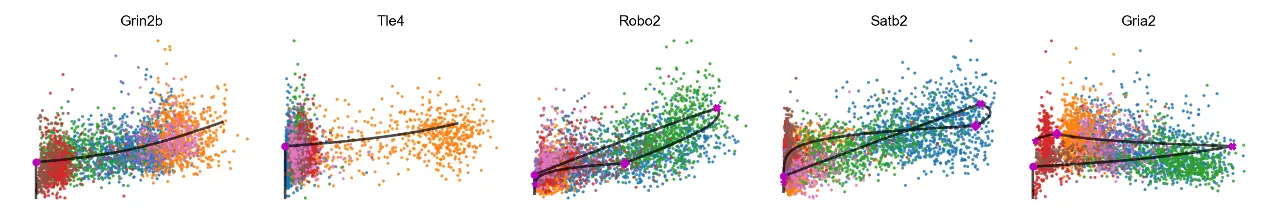
# 3D phase portraits, colored by celltype.
mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='cus', axis_on=False, downsample=2)
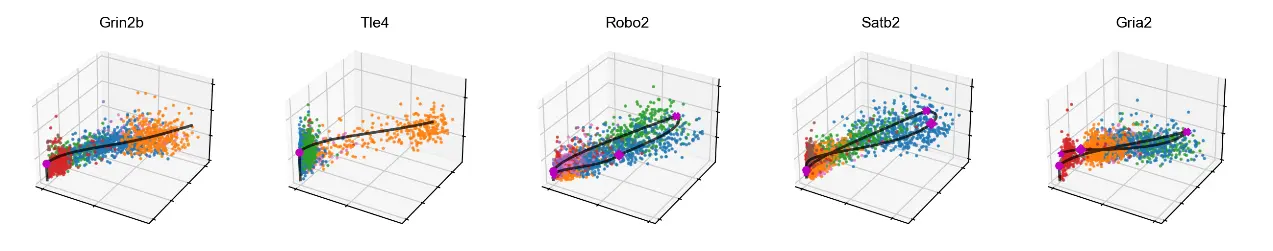
We can also add velocity arrows to phase portraits to show the predicted directions
mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='us', axis_on=False, frame_on=False, downsample=2, velocity_arrows=True)

mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='cu', axis_on=False, frame_on=False, downsample=2, velocity_arrows=True)
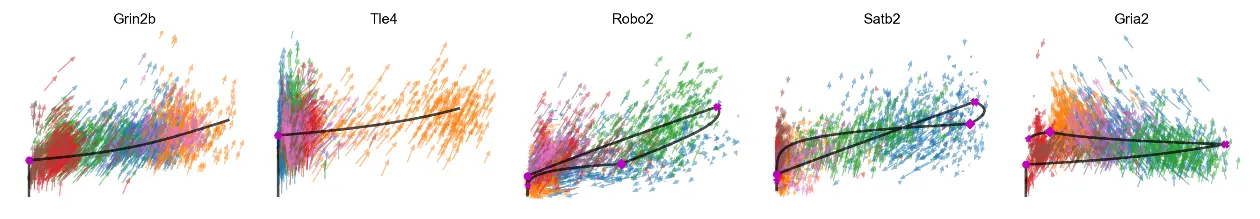
mv.scatter_plot(adata_result, gene_list, color_by='celltype', by='cus', downsample=3, velocity_arrows=True)
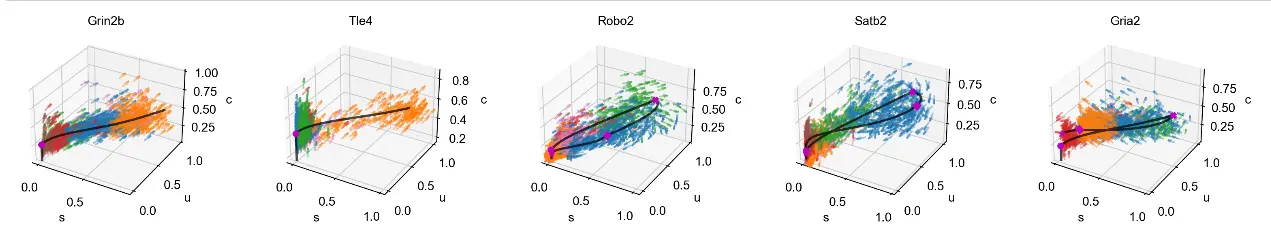
Github在MultiVelo
示例,小鼠脑、文章数据
生活很好,有你更好

















 38
38

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









