







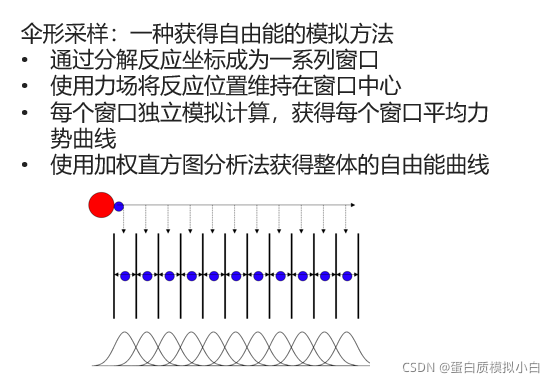
 本文提供了一份详细的GROMACS伞形采样模拟教程,涵盖从准备拓扑文件到数据分析的整个过程。通过GROMACS进行蛋白质结合自由能计算,涉及能量最小化、溶剂和离子添加、构型生成、伞形采样模拟及数据处理等步骤。使用GROMACS进行分子动力学模拟,并借助北鲲云计算平台加速计算。
本文提供了一份详细的GROMACS伞形采样模拟教程,涵盖从准备拓扑文件到数据分析的整个过程。通过GROMACS进行蛋白质结合自由能计算,涉及能量最小化、溶剂和离子添加、构型生成、伞形采样模拟及数据处理等步骤。使用GROMACS进行分子动力学模拟,并借助北鲲云计算平台加速计算。
GROMACS教程:伞形采样|Jerkwin参考教程如下,准备计算蛋白质结合的自由能。
新建文件夹 opt/work5 做为计算文件夹
第一步: 准备拓扑文件
下载2BEG文件,放在work5文件夹,重命名为input.pdb
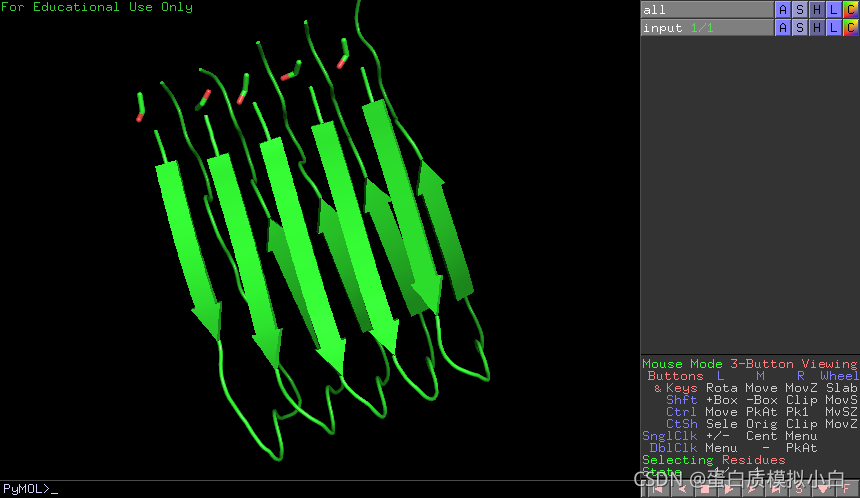
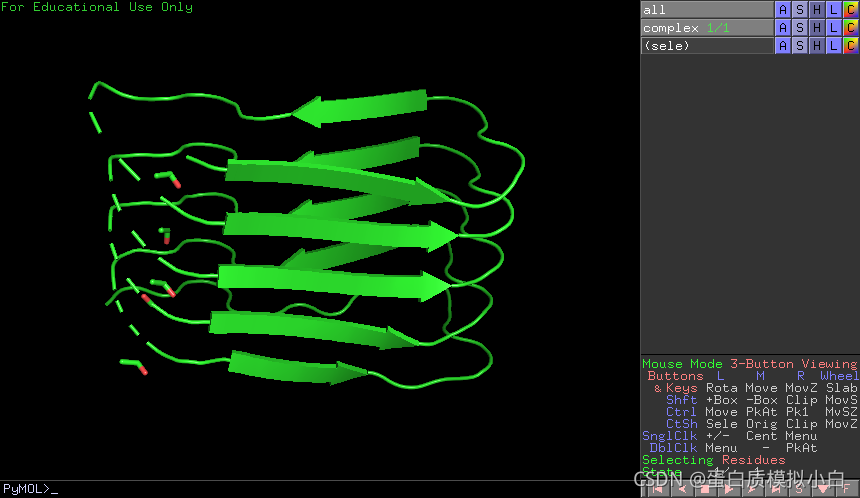
pymol里看起来是这样的,Pymol可以官网免费下载教育版。
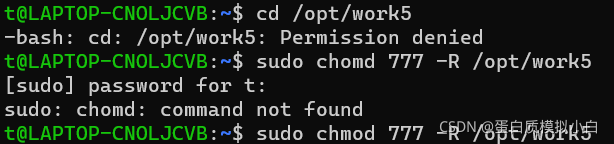
先改文件夹权限,方便后面不会permission denied
sudo chmod 777 -R /opt/work5
使用gmx pdb2gmx处理结构:
gmx pdb2gmx -f input.pdb -ignh -ter -o complex.gro
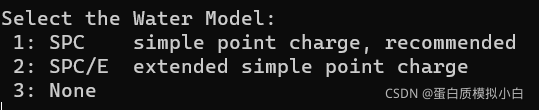
选水模型 选1
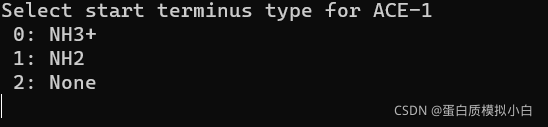
选N端 选2
下一个选 COO-
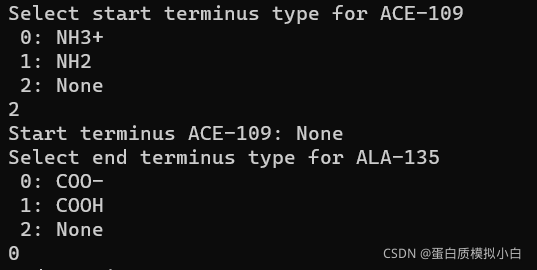
会选很多次,选错会报错。
现在的文件
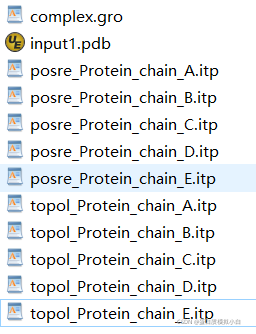
在topol_Protein_chain_B.itp文件的末尾加上下面几行:
#ifdef POSRES_B
#include "posre_Protein_chain_B.itp"
#endif看一下complex文件
第二步: 定义单元晶胞
gmx editconf -f complex.gro -o newbox.gro -center 3.280 2.181 2.4775 -box 6.560 4.362 12
第三步: 添加溶剂和离子
gmx solvate -cp newbox.gro -cs spc216.gro -o solv.gro -p topol.top
加完溶液的样子
添加离子 使用ions.mdp文件
; ions.mdp - used as input into grompp to generate ions 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 602
602

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









