









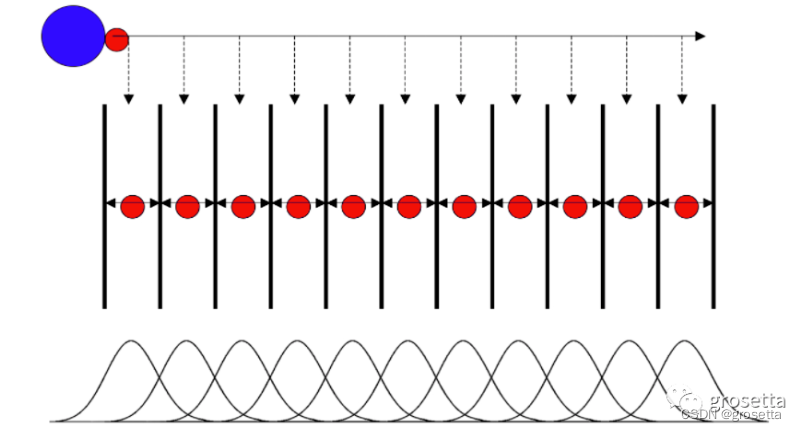
伞形采样是一种加速采样方法。对于生物大分子而言,一些大的构象变化在现有的模拟尺度范围内很难达到或者根本无法达到。这种情况下人们往往采用给体系添加外部偏置力的手段加速这一过程的发生。具体的就是在大分子某一部位施加一个简谐力(可以想象成弹簧),以恒定速度牵引其移动。这一过程属于拉伸动力学(SMD)范畴。在拉伸过程中,上述被牵引的区域发生位移,然后挑选这期间的一些构象,以这些构象为初始帧进行独立的模拟(如图1)。最后用加权直方的手段对这些模拟进行整合,得出牵引过程能量变化情况。这一过程叫伞形采样。由此可见拉伸动力学模拟和伞形采样是密不可分的。本文将以溶菌酶配体复合物为例演示这一过程,拉伸结果见图2。
下文需要用到的各类文件wx后台回复“伞形采样” 自取。
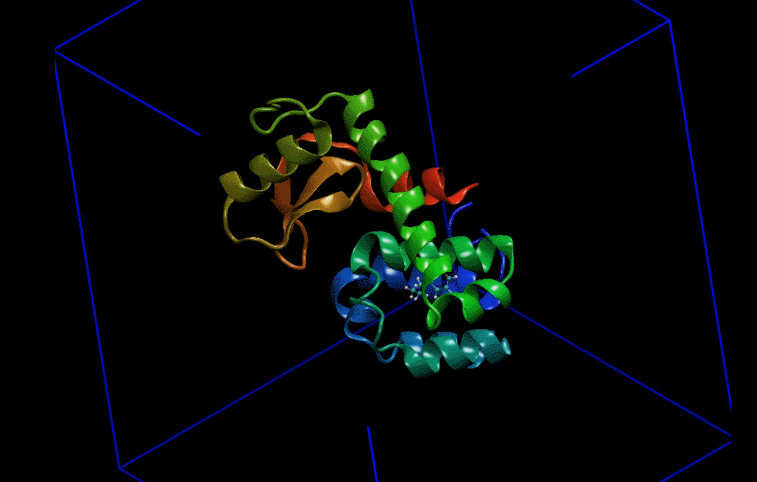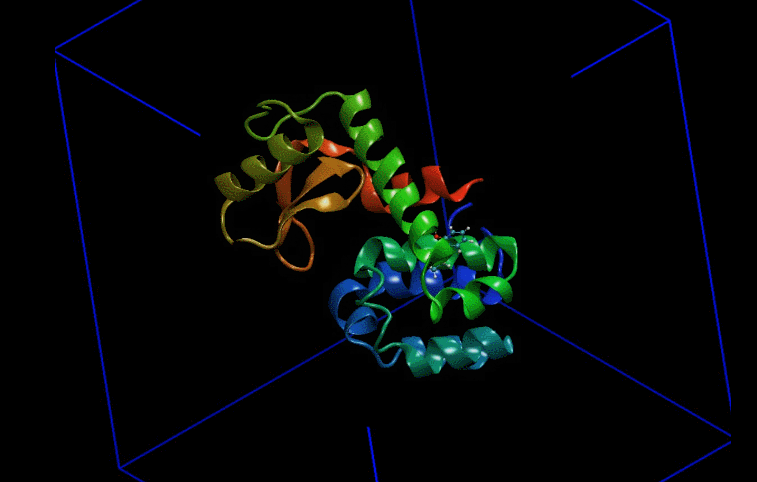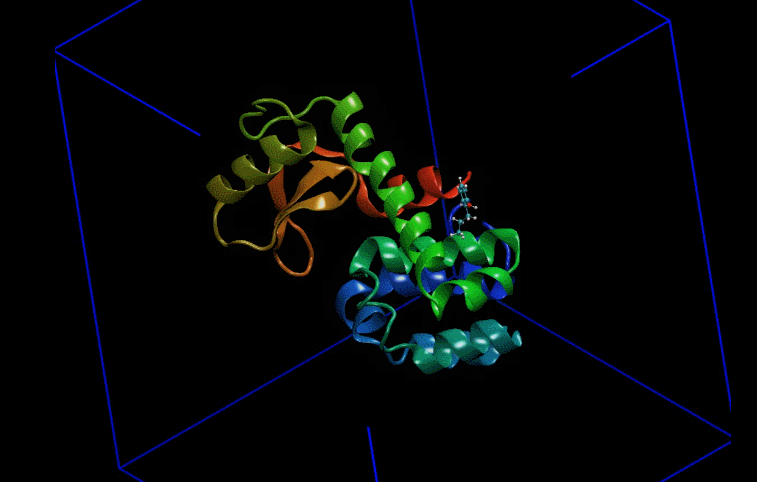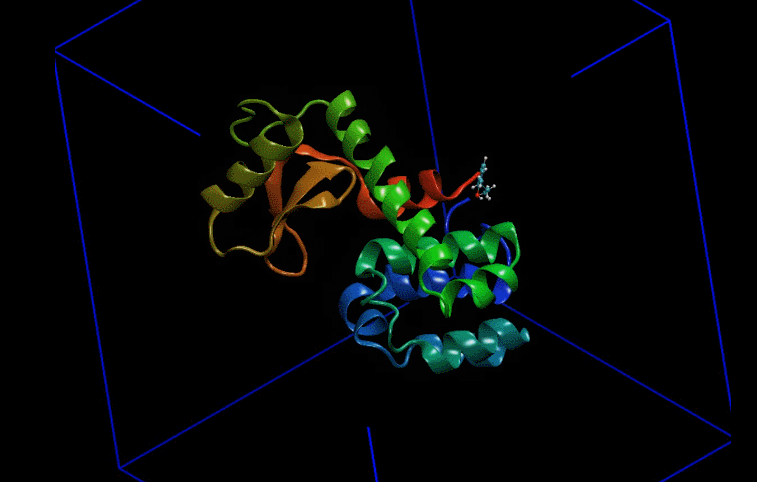
1.常规模拟
这一步可以参照Gromacs官网(http://www.mdtutorials.com/gmx/complex/index.html)溶菌酶复合物教程。
可以跟着做到获得NPT模拟终状态为止。
当然你也可以直接使用提供的示例文件
2.拉伸模拟
这一步和常规成品模拟有两点区别:
1、制作索引基团的index文件:
比如我想要牵引的两个对象分别是配体和蛋白,那么需要将二者各自存放在一组内。本例中它们已被gromacs自动识别,所以无需新建。
cd pull #进入示例文件夹
gmx make_ndx -f NPT.gro -o index.ndx
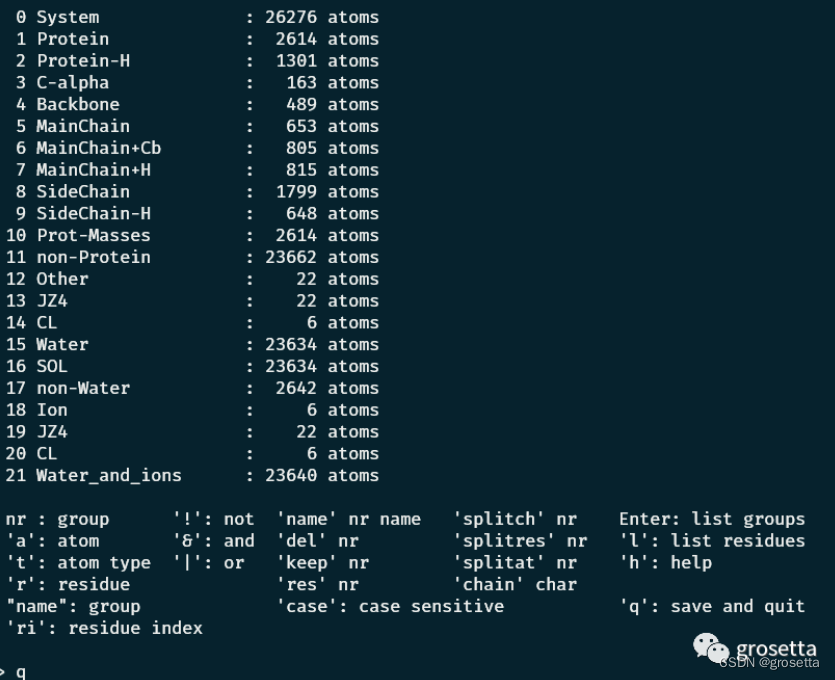
记住蛋白所在组和配体所在组的组名,分别是Protein、JZ4。然后输入q,回车退出。
2、需要在成品模拟参数文件(mdp文件)基础上添加牵引“控件”:
如下所示:
pull = yes ; 开启牵引
pull_ncoords = 1 ; 设置有几个拉伸反应过程
pull_ngroups = 2 ; 在一个拉伸过程涉及两个拉伸组
pull_group1_name = JZ4 ; 第一个拉伸组的名称叫做JZ4,对应index文件的配体组名
pull_group2_name = Protein ; 第二个拉伸组的名称叫做Protein, 对应index文件的蛋白组名
pull_coord1_type = umbrella ; 使用简谐力牵引(类似于弹簧)
pull_coord1_geometry = direction-periodic ; 牵引模式,有以下四种类型
; distance:沿质心间的矢量, 用pull-dim选择分量
; direction:沿pull-vec方向
; direction-periodic:同direction, 距离可超过盒长一半. 使用时关闭压力耦合
; cylinder:圆柱
pull_coord1_dim = N N Y ; 沿着Z轴方向牵引,和distance牵引模式搭配使用
pull-coord1-vec = 0 0 1 ; 沿着(0,0,1)矢量方向牵引,和direction、direction-periodic两种牵引模式搭配使用
pull_coord1_groups = 1 2 ; groups1 对应上面的JZ4, groups2对应Protein
pull_coord1_start = yes ; 定义初始质心距离大于0
pull_coord1_rate = 0.05 ; 牵引速度0.05nm/ps
pull_coord1_k = 1000 ; 牵引力常数,单位(kJ mol^-1 nm^-2)
;}==============================================================================
执行牵引模拟
gmx grompp -f pull.mdp -c NPT.gro -r NPT.gro -n index.ndx -o pull.tpr
#注意上一步可能在gmx2019及以后的版本会报错,这是因为19版以后有新的参数添加,18版本可以通过。\
#如果您不想额外安装18版gmx,可以尝试了解一下pull-pbc-ref-prev-step-com 以及 pull_group2_pbcatom这两个参数。
gmx mdrun -deffnm pull -v
3.伞形采样
cd ../umbrella_sample/ #定位到伞形采样目录下
等时间间隔抽取关键帧并执行批量独立模拟,这一步脚本可以代替:
./run.sh
run.sh脚本内容如下:
#!/bin/bash
for i in {400..1000}; #脚本循环数
do
t=$(awk -v i=$i 'BEGIN{print 0.05*i}') #0.05表示抽取的时间间隔(ps)
#本例运行了50ps,因此最多有1000个间隔,循环数不能超过1000
#这些参数可以根据自己的需求更改,这只是示例,随便设的。
name=pull_$t
echo 0 | gmx trjconv -s ../pull/pull.tpr -f ../pull/pull.xtc -dump $t -o $name.gro
gmx grompp -f md.mdp -p ../pull/topol.top -c $name.gro -o $name.tpr -maxwarn 99
gmx mdrun -s $name.tpr -pf ${name}_pf.xvg
rm -f *.edr *.log *.trr *.cpt *.xtc *.gro
rm \#*
done
脚本可能需要根据具体需求调整参数的部分,详见注释。
注意,此步脚本中用到的mdp参数文件需要将牵引代码控制部分的牵引速度设为0,在提供的示例文件里也可以查看。
接下来,需要将路径中新生成的tpr文件以及pf.xvg文件名保存到各自的列表文件中,如下:
ls *.tpr | awk -F '.t' '{print $1}' | sort -t '_' -nk 2 | sed "s/$/.tpr/g" > tpr_files.dat
ls *_pf.xvg | awk -F '.x' '{print $1}' | sort -t '_' -nk 2 | sed "s/$/.xvg/g" > pullf_files.dat
4. 分析
执行wham(加权直方分析)程序计算PMF(平均力势)。
gmx wham -it tpr_files.dat -if pullf_files.dat -o hist -unit kCal -b 1 -e 8
伞形采样选取的一系列初始构象是拉伸过程中的某一帧,它们都处于非平衡态,因此对于这些独立的模拟在进行加权分析时往往需要舍弃前几百ps不平衡的轨迹,也就是上条命令中的-b参数控制的。-b 1 和 -e 8意思是只处理每条轨迹1ps~8ps之间的部分。
由于伞形采样计算量巨大,所以我在测试时将每条模拟轨迹缩减到8ps,好在结果还看得过去。
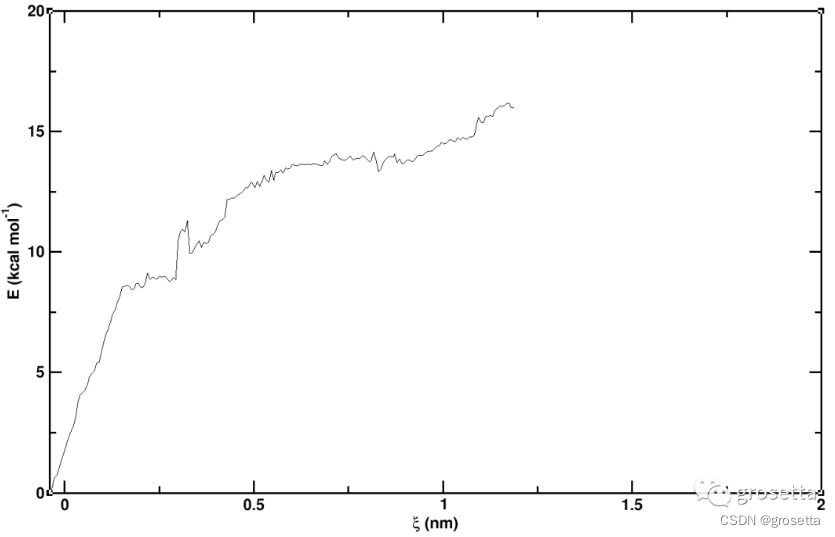
上图最高点和最低点的差值大约等于被拉伸分子的结合能ΔG(kcal/mol)。
值得注意的是,上文提到拉伸过程中施加的力是简谐力,其特点是当作用到拉伸物体上时,被拉伸物与施加力起点存在运动不同步现象,这主要取决于“弹簧”的弹性系数,如果弹性系数越强二者运动越同步。这可以和中学经典问题“弹簧拉动滑块”类比。另外被拉伸物运动方向不一定和力的施加方向一致,因为被拉伸物体(原子)还会受到周围原子的阻碍,它会曲曲绕绕地朝着大致的方向运动。因此这也可以被用来研究分子最优解离路径问题。
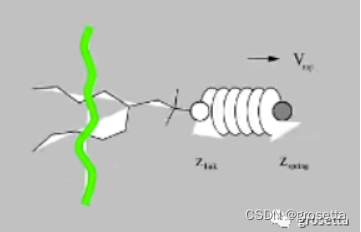
如果时间充足的话,强烈建议您在拉伸过程中可以将力常数设得小些。这样也会大大有利于后续伞形采样分析。


















 602
602

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











