







 这篇博客记录了gromacs初学者在学习过程中遇到的'Atom index n in position_restraints out of bounds'错误及其解决方案。问题源于topol.top文件中分子位置约束文件的不正确排列,正确做法是将各部分的itp和posre.itp文件合并。强调了topol.top文件的编写正确性对避免此类错误的重要性。
这篇博客记录了gromacs初学者在学习过程中遇到的'Atom index n in position_restraints out of bounds'错误及其解决方案。问题源于topol.top文件中分子位置约束文件的不正确排列,正确做法是将各部分的itp和posre.itp文件合并。强调了topol.top文件的编写正确性对避免此类错误的重要性。
gromacs学习中,面临各种报错,作为初学者,自己把踩过的坑一一记录下来,希望对后来者有所帮助。
1. 报错: Atom index n in position_restraints out of bounds
解决方案:见官网,链接:http://www.gromacs.org/Documentation/Errors#Atom_index_n_in_position_restraints_out_of_bounds
解释:
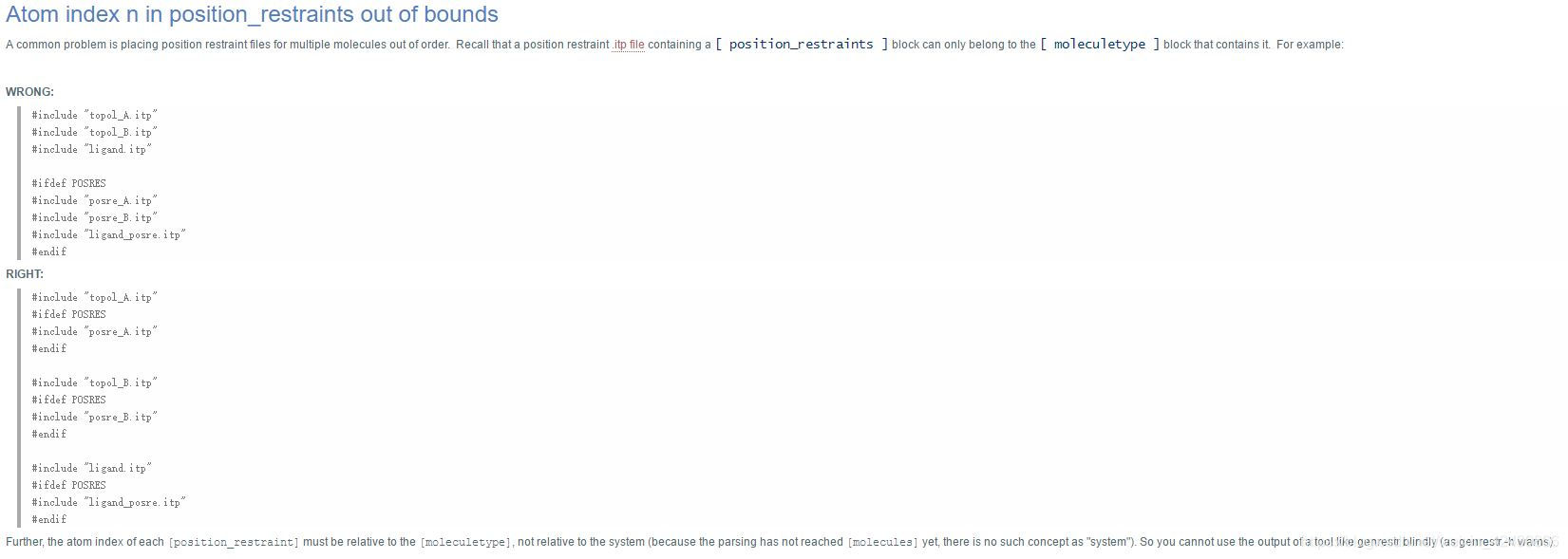
简单说就是topol.top文件中最后部分分子的的位置约束文件排列不整齐,比如:
WRONG:
#include “topol_A.itp”
#include “topol_B.itp”
#
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1370
1370

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









